Im Gegensatz zu Halbleitern unterscheiden sich Isolatoren wegen der großen
Energielücke zwischen Valenz- und Leitfähigkeitsband. Im allgemeinen werden
auch bei hohen Temperaturen nur wenig Elektronen im Leitfähigkeitsband
anzutreffen sein. Berechnungen mit der Pseudopotentialmethode [117] und
einem sogenannten tight-binding-Verfahren [118] ergeben eine
Energiedifferenz von ungefähr 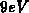 zwischen diesen beiden Bändern für
zwischen diesen beiden Bändern für
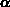 -Quarz in einer hexagonalen Struktur. Die Brillouinzone einer hexagonal
flächenzentrierten Struktur ist in Abbildung 3.9 eingezeichnet. Das
direkte Gitter besteht aus drei Silizium- und sechs Sauerstoffatomen
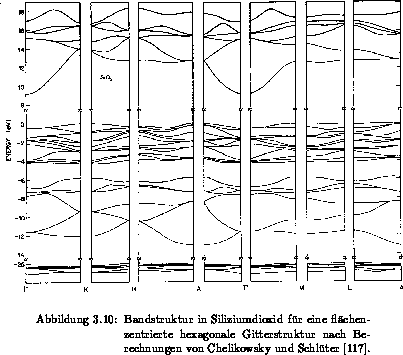
[119]. Abbildung 3.10 zeigt die einzelnen Bänder in
Siliziumdioxid, die mit der Pseudopotentialmethode berechnet worden sind
[117]. Der Nullpunkt entspricht dem Maximum des höchsten Valenzbandes.
Das erste Leitfähigkeitsband erstreckt sich über
-Quarz in einer hexagonalen Struktur. Die Brillouinzone einer hexagonal
flächenzentrierten Struktur ist in Abbildung 3.9 eingezeichnet. Das
direkte Gitter besteht aus drei Silizium- und sechs Sauerstoffatomen
[119]. Abbildung 3.10 zeigt die einzelnen Bänder in
Siliziumdioxid, die mit der Pseudopotentialmethode berechnet worden sind
[117]. Der Nullpunkt entspricht dem Maximum des höchsten Valenzbandes.
Das erste Leitfähigkeitsband erstreckt sich über 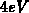 und wird somit den
Elektronentransport in Siliziumdioxid wesentlich bestimmen. Die Anisotropie
dieses Bandes, das im Mittelpunkt der Brillouinzone lokalisiert ist, kann als
gering angesehen werden [109][110]. Da Siliziumdioxid als
Gate-Isolator eine amorphe Struktur aufweist, wird in [120][121]
darauf hingewiesen, daß die elektronischen Eigenschaften einer
und wird somit den
Elektronentransport in Siliziumdioxid wesentlich bestimmen. Die Anisotropie
dieses Bandes, das im Mittelpunkt der Brillouinzone lokalisiert ist, kann als
gering angesehen werden [109][110]. Da Siliziumdioxid als
Gate-Isolator eine amorphe Struktur aufweist, wird in [120][121]
darauf hingewiesen, daß die elektronischen Eigenschaften einer
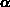 -Quarzstruktur sehr ähnlich sind. Die Zustandsdichte in Siliziumdioxid
weist zwischen
-Quarzstruktur sehr ähnlich sind. Die Zustandsdichte in Siliziumdioxid
weist zwischen 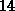 und
und 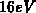 ein scharfes Minimum auf. In Berechnungen von
Titaniumdioxid, das strukturelle und kristallspezifische Ähnlichkeiten
mit Siliziumdioxid hat, ist die Zustandsdichte für das Leitfähigkeitsband
eingezeichnet [122]. Ein Minimum ist dort genau dann festzustellen, wenn
innerhalb der Bandstruktur eine Lücke auftritt.
ein scharfes Minimum auf. In Berechnungen von
Titaniumdioxid, das strukturelle und kristallspezifische Ähnlichkeiten
mit Siliziumdioxid hat, ist die Zustandsdichte für das Leitfähigkeitsband
eingezeichnet [122]. Ein Minimum ist dort genau dann festzustellen, wenn
innerhalb der Bandstruktur eine Lücke auftritt.
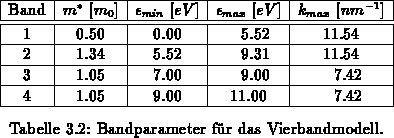
Zur Berechnung der Bandstruktur [123] wird einerseits ein sphärisch
parabolisches,
ein sphärisch nichtparabolisches als auch ein analytisches Vierbandmodell
[58] verwendet. In Tabelle 3.2 sind die einzelnen
Parameter angegeben. Für das nichtparabolische Band beträgt die
Nichtparabolizität 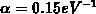 . Die Energierelationen in
Abhängigkeit des Wellenvektors lauten
. Die Energierelationen in
Abhängigkeit des Wellenvektors lauten
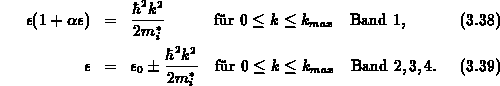
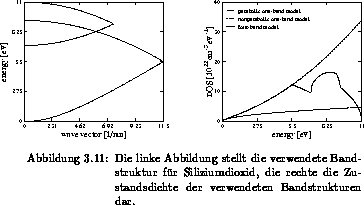
Das Vierbandmodell und die dazugehörige Zustandsdichte ist in Abbildung
3.11 dargestellt und mit einem parabolischen und nichtparabolischen
Einbandmodell verglichen. Mit der Wahl des ersten Bandes ist das zweite
eindeutig festgelegt [58]. Die Wahl des dritten und vierten Bandes
erfolgt mit einer numerischen Approximation der einzelnen Bänder der vollen
Bandstruktur, wobei die Tatsache berücksichtigt wird, daß bei 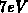 ein
Minimum auftritt. Für das dritte und vierte Band sind einfache Berechnungen zur
Reproduktion der tatsächliche Zustandsdichte ausgeführt worden.
ein
Minimum auftritt. Für das dritte und vierte Band sind einfache Berechnungen zur
Reproduktion der tatsächliche Zustandsdichte ausgeführt worden.
Experimentelle Arbeiten [124][125], bei denen die Streurate
extrahiert worden ist, lassen Rückschlüsse auf die Zustandsdichte zu. Ein
Minimum der Streurate wird zwischen 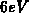 und
und 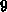 eV festgestellt. Ein zweites
Maximum ist erst bei sehr hohen Energien (
eV festgestellt. Ein zweites
Maximum ist erst bei sehr hohen Energien (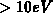 ) angegeben
) angegeben . Dieses Maximum kann für elektrische
Feldstärken unter
. Dieses Maximum kann für elektrische
Feldstärken unter 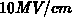 vernachlässigt werden. In [124] liegt das
Hauptaugenmerk auf Elektronen mit Energien von mehr als
vernachlässigt werden. In [124] liegt das
Hauptaugenmerk auf Elektronen mit Energien von mehr als 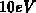 . Es wird ein tief
inelastischer Streuprozeß postuliert, der bei Elektronen für einen
Energieverlust von ungefähr
. Es wird ein tief
inelastischer Streuprozeß postuliert, der bei Elektronen für einen
Energieverlust von ungefähr 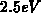 verantwortlich ist. Als Erklärung für
diesen Prozeß wird Elektron-Elektronwechselwirkung vorgeschlagen. Eine
detaillierte Untersuchung dieses Streumechanismus' ist ausständig.
verantwortlich ist. Als Erklärung für
diesen Prozeß wird Elektron-Elektronwechselwirkung vorgeschlagen. Eine
detaillierte Untersuchung dieses Streumechanismus' ist ausständig.