As proposed by Slater in 1954, the bandstructure of silicon (as well as that of other diamond-like semiconductors) can be calculated using the ![]() tight-binding method. This method considers four valence orbitals
tight-binding method. This method considers four valence orbitals ![]() and
and ![]() , including
, including ![]() ,
, ![]() , and
, and ![]() , for each silicon atom [31]. To enhance accuracy for reproducing the conduction band of diamond and zinc blende semiconductors, it is common to use the
, for each silicon atom [31]. To enhance accuracy for reproducing the conduction band of diamond and zinc blende semiconductors, it is common to use the ![]() tight-binding method in which the first excited
tight-binding method in which the first excited ![]() -like orbital
-like orbital ![]() (i.e.
(i.e. ![]() in the case of silicon) is also included [46]. It was further improved by including the five excited
in the case of silicon) is also included [46]. It was further improved by including the five excited ![]() orbitals
orbitals ![]() in the so called
in the so called
![]() tight-binding method. This method includes ten orbitals per silicon atom.
tight-binding method. This method includes ten orbitals per silicon atom.
We first consider the
![]() tight-binding method without spin-orbit coupling for bulk silicon, the unitcell of which consists of two atoms commonly called the anion and the cation. Each anion atom is connected to four cation atoms through for bonds
tight-binding method without spin-orbit coupling for bulk silicon, the unitcell of which consists of two atoms commonly called the anion and the cation. Each anion atom is connected to four cation atoms through for bonds ![]() ,
, ![]() ,
, ![]() , and
, and ![]() and vice versa. The Hamiltonian of the unitcell, when considering the coupling of the anion with only one neighboring cation, consists of four blocks as:
and vice versa. The Hamiltonian of the unitcell, when considering the coupling of the anion with only one neighboring cation, consists of four blocks as:
![$\displaystyle H= \left[ \begin{array}{ll} H_{aa} & H_{ac} \\ H_{ca} & H_{cc} \\ \end{array} \right]$](img183.png) |
(2.13) |
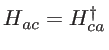 the coupling between two atoms. In the case of silicon, we note that
the coupling between two atoms. In the case of silicon, we note that
| (2.14) |
| (2.16) |
| (2.17) |
To describe the top of the valence band correctly, one needs to include the spin-orbit coupling [32]. In this case, the number of orbitals per atom increases to 20 [44,47]. However, the spin-orbit interactions affect only orbitals with different spins of the same atom. Therefore, they are only added to the diagonal blocks ![]() , and
, and ![]() of the Hamiltonian [47].
of the Hamiltonian [47].
![$\displaystyle H_{ac}=[g_{i,j}^B V_{i,j}^{ac}]$](img189.png)