
 |
|
||||
BiographyDiego Milardovich was born in Rosario, Argentina in 1992. He received his Master's degree in Electronic Engineering at the National University of Rosario in 2019. In February 2020, he joined the Institute for Microelectronics at the TU Wien, where he is currently working on his doctoral degree focusing on atomistic modeling of insulator defects. His previous experiences include 1 year as a research assistant at the Neuroscience and Biomedical Engineering department of Aalto University (Helsinki, Finland), 2 years as an engineering officer at Nestlé (Firmat, Argentina) and 3 months as a research engineer at Techint (Buenos Aires, Argentina). |
|||||
Building Robust Machine Learning Force Fields by Composite Gaussian Approximation Potentials
It has become increasingly necessary in recent years to include atomistic calculations in simulation workflows of nanoelectronic devices. This translates into a growing interest in machine learning (ML) interatomic potentials, motivated by their relatively high accuracy and remarkably low computational costs when compared to ab-initio methods, such as density functional theory (DFT). While easy access to a wide range of ML software packages facilitates the implementation of new potentials, the development of robust ML potentials is still a topic of active research in the field. One of the main obstacles which weakens the robustness of these potentials is overfitting, an undesired statistical phenomenon by which ML models perform well on training data but poorly in real test scenarios.
In this work, we developed a systematic solution to mitigate the effects of overfitting. We proposed to augment traditional interatomic potentials by adding a set of lower-dimensional auxiliary ML models, as shown in Fig. 1(a). The proposed solution is demonstrated by building an ML interatomic potential for amorphous silicon dioxide (SiO2), using the Gaussian approximation potential (GAP). GAP is a kernel-based method, which assumes that similar local atomic environments give similar contributions to the total potential energy of a given atomic system. The input atomic configuration is compared to those in a training dataset by means of a kernel function, and its energy contribution is computed as the weighted sum of these similarity measurements, as shown in Fig. 1(b).
The ML interatomic potential built with our proposed approach includes an auxiliary potential to account for short-range atomic interactions, and it was validated against a traditional ML interatomic potential (i.e., composed of a single GAP). Both potentials were first tested by using them to compute the potential energies of a set of 1000 amorphous SiO2 structures and comparing their results against DFT, both having a virtually identical accuracy. These results are presented in Fig. 2(a). The next test involved using both potentials to run the exact same melt-and-quench molecular dynamics (MD), as presented in Fig. 2(b). The results of these MDs are shown in Figs. 2(c)-(e), where unphysical behavior (in the form of clustering) can be seen in the amorphous structures created with the traditional potential. However, this is not present in the amorphous structures created with our proposed composite potential. This becomes clear when analyzing the radial density functions (RDFs) in Fig. 2(f).
The results indicate that the addition of these auxiliary ML models increases the robustness of the overall ML interatomic potential. Moreover, these benefits come at a negligible increase in computational costs.
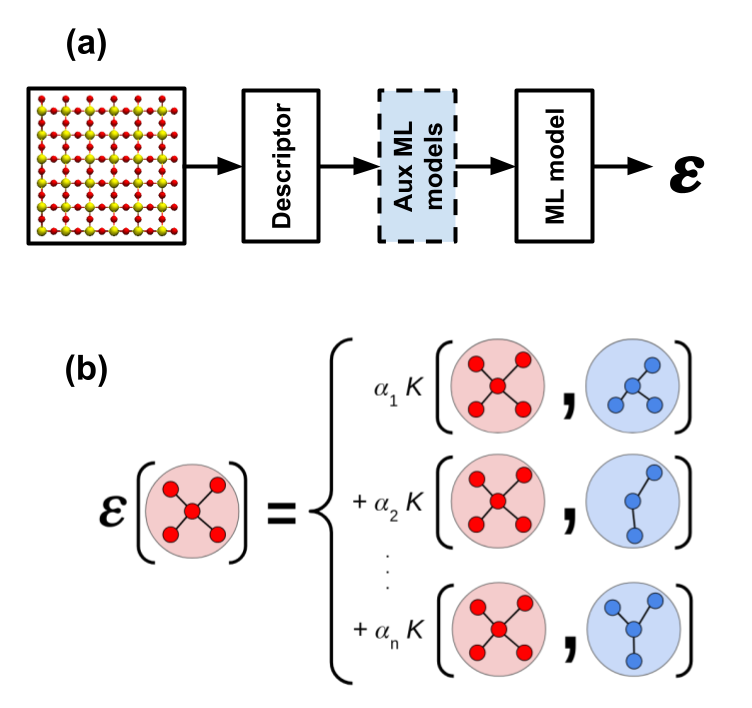
Fig. 1: (a) Proposed framework to build ML interatomic potentials. A traditional ML interatomic potential is composed of a descriptor (i.e., a mathematical representation of a given atomic system) and a single ML model to compute the potential energy from the descriptor. We propose to include a set of lower-dimensional auxiliary ML models to reduce overfitting. (b) Schematic of a GAP. This type of kernel-based method compares the input atomic configuration (red) to those in a training dataset (blue) by means of a kernel function (K). The potential energy is then computed as the weighted sum of these similarity measurements.
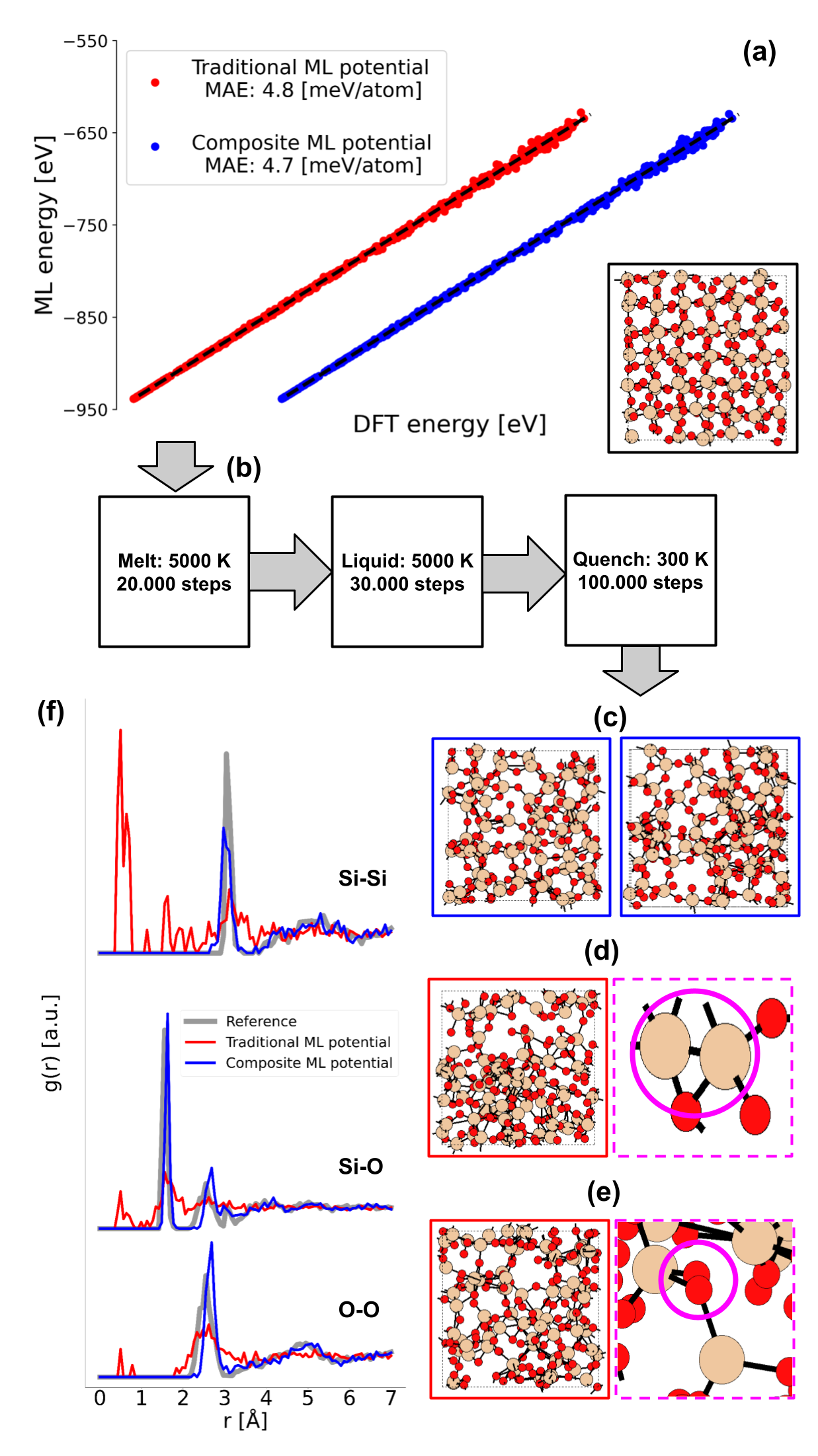
Fig. 2: Validating our composite ML potential against a traditional ML potential. (a) Comparing their energy estimations against DFT for a set of 1000 amorphous SiO2 structures. (b) Using both potentials to run melt-and-quench MDs. (c) Amorphous structures obtained with our composite potential. (d) and (e) Amorphous structures obtained with the traditional ML potential, together with examples of unphysical behavior found in them. (f) Comparing the RDFs of the amorphous structures obtained with both potentials against the DFT reference.