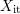 [56, 57, 58] in
[56, 57, 58] in
In the standard reaction-diffusion theory the kinetic rate equation describing the
interface reaction via a hydrogen species [56, 57, 58] in
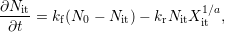 | (C.1) |
with 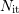 and
and 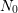 as unpassivated and total amount of interface states. Thus
as unpassivated and total amount of interface states. Thus
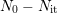 denotes the concentration of passivated interface defects depassivating
with the rate
denotes the concentration of passivated interface defects depassivating
with the rate 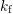 . The passivation rate
. The passivation rate 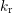 of the dangling bonds also depends
on the hydrogen species
of the dangling bonds also depends
on the hydrogen species 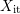 with its kinetic exponent
with its kinetic exponent  (
(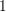 for
for 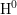 and
and 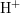 ,
and
,
and  for
for 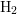 ) [172]. Assuming the quasi-equilibrium regime of the interface
reaction (
) [172]. Assuming the quasi-equilibrium regime of the interface
reaction (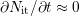 ) as the dominant regime after [59, 66, 17, 71], the rate
equation (C.1) can be rewritten as
) as the dominant regime after [59, 66, 17, 71], the rate
equation (C.1) can be rewritten as
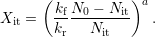 | (C.2) |
The boundary value problem for 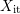 is as follows:
is as follows:
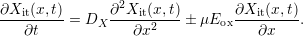 | (C.3) |
When neglecting charged hydrogen (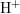 ), the drift term inside
the drift-diffusion process (C.3) vanishes. The remaining diffusion
process will be approximately solved on basis of a triangular hydrogen
profile1
[85, 59], as depicted in Fig. C.1 (right).
), the drift term inside
the drift-diffusion process (C.3) vanishes. The remaining diffusion
process will be approximately solved on basis of a triangular hydrogen
profile1
[85, 59], as depicted in Fig. C.1 (right).
After the continuity equation the interface states additionally created,
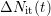 , are due to the leaving hydrogen
, are due to the leaving hydrogen 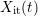 , which is shown in Fig. C.1
(middle). The corresponding diffusion front is given by
, which is shown in Fig. C.1
(middle). The corresponding diffusion front is given by 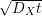 . Comparing with
Fig. C.1 (right) yields the area confined by the diffusion front on the one
hand and the number of
. Comparing with
Fig. C.1 (right) yields the area confined by the diffusion front on the one
hand and the number of 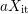 at the interface at the other hand which
equals
at the interface at the other hand which
equals
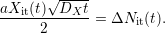 | (C.4) |
The ratio between the diffusing hydrogen species and resulting interface states is
determined by the kinetic exponent  , i.e. each
, i.e. each 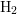 leaves
leaves  dangling
bonds.
dangling
bonds.
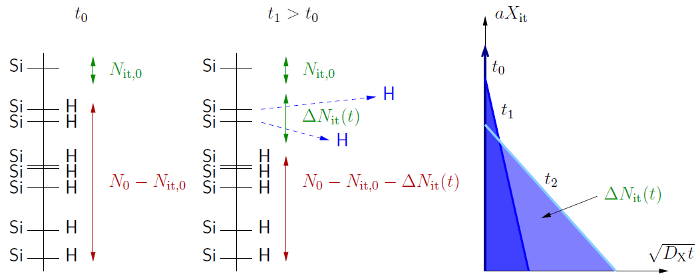
To solve equations (C.2) and (C.4) the following assumptions are made: (i)
The amount of passivated interface states 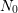 is much larger than the initial
value of
is much larger than the initial
value of 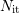 ,
, 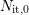 , and (ii)
, and (ii) 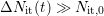 . A schematic picture of
the interface shows all necessary quantities and is relations (Fig. C.1
(left)). Inserting these assumptions into (C.2) and comparing with (C.4)
gives
. A schematic picture of
the interface shows all necessary quantities and is relations (Fig. C.1
(left)). Inserting these assumptions into (C.2) and comparing with (C.4)
gives
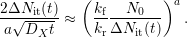 | (C.5) |
The approximated number of 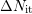 is then
is then
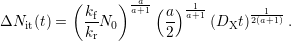 | (C.6) |
By using atomic hydrogen (a=1) this term simplifies to
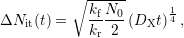 | (C.7) |
while molecular hydrogen (a=2) yields
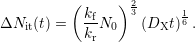 | (C.8) |
Alternatively (C.4) can be formulated via the flux of the hydrogen profile (the gradient right at the interface) and yields a first-order differential equation in time to solve. The results differing by a constant prefactor from the algebraic expressions in (C.6) are summarized in [71].
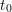 , i.e.
, i.e. 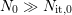 .
The additionally created dangling bonds
.
The additionally created dangling bonds  are furthermore assumed to
dominate the total number of dangling bonds (
are furthermore assumed to
dominate the total number of dangling bonds (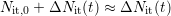 ).
Note that in fact the
).
Note that in fact the 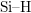 and the silicon dangling bonds are not arranged
in two groups like schematically depicted here, but are randomly distributed.
Right: Hydrogen profile inside the oxide during a diffusion process. The
area under the hydrogen profile with its progressing diffusion front
and the silicon dangling bonds are not arranged
in two groups like schematically depicted here, but are randomly distributed.
Right: Hydrogen profile inside the oxide during a diffusion process. The
area under the hydrogen profile with its progressing diffusion front 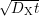 equals the number of additionally generated interface states
equals the number of additionally generated interface states 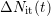 given
by relation (C.4).
given
by relation (C.4).