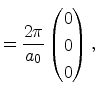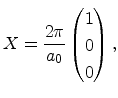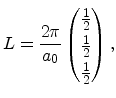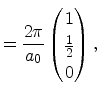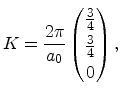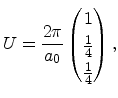|
|
|
|
Previous: 3.3.3 Strain Resulting from Epitaxy Up: 3. Strained Bulk Band Structure Next: 3.4.1 Band Structure of Relaxed Si |
The structure depicted in Figure 3.4 consists of two basis atoms and may be
thought of as two inter-penetrating face centered cubic (fcc) lattices, one
displaced from the other by a translation of
![]() along a body diagonal.
along a body diagonal.
[a]![\includegraphics[width=2.6in]{inkscape/DiamondLattice.eps}](img238.png) [b]
[b]![\includegraphics[width=2.45in]{inkscape/DiamondLattice2.eps}](img239.png)
|
In cubic semiconductors such as Si or Ge the two atoms of the basis are identical and the structure is called the diamond structure. If the two basis atoms are different, the structure is called the zinc-blende structure. Many III-V semiconductors such as GaAs, AlsAs, InAs, or InP are of zinc-blende type.
In Figure 3.4 the diamond structure is depicted. The primitive basis
vectors and the two atoms at ![]() and
and
![]() are
highlighted in Figure 3.4b. The basis vectors of the direct
Bravais lattice are
are
highlighted in Figure 3.4b. The basis vectors of the direct
Bravais lattice are
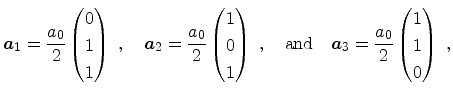 |
(3.21) |
| (3.22) |
The basis vectors of the reciprocal lattice
![]()
![]() are obtained from the relation
are obtained from the relation
![]() as
as
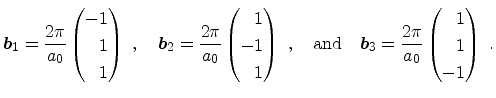 |
(3.23) |
The first Brillouin zone (BZ) represents the central (Wigner-Seitz) cell of the reciprocal lattice. It contains all points nearest to the enclosed reciprocal lattice point. The boundaries of the first BZ are determined by planes which are perpendicular to the reciprocal lattice vectors pointing from the center of the cell to the 14 lattice points nearest to the origin of the cell at their midpoints. The 14 faces are
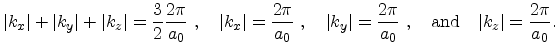 |
(3.25) |
[a]![\includegraphics[width=2.6in]{inkscape/bz.eps}](img253.png) [b]
[b]![\includegraphics[width=2.6in]{inkscape/bzOctand.eps}](img254.png)
|
Due to the translational invariance of the lattice the wave functions and the energy bands are periodic in the reciprocal space and it is sufficient to consider only the first BZ for band structure calculations [Yu03].
The diamond structure is invariant not only under translations, but also under
several other symmetry operations such as reflections, rotations, or inversion.
These symmetry operations are usually denoted as point operations, since they
leave at least one point of the lattice invariant, which is not the case for
translations. The set of all point operations for a particular crystal
structure forms a group which is denoted as point group. The point group of the
diamond structure has 48 symmetry elements which are reflected in the symmetry
of the first BZ. A quick examination (see Figure 3.5) shows that the BZ is
invariant under various rotations, for example 90![]() rotations about the
rotations about the
![]() ,
,
![]() , and
, and
![]() axes and under reflections through certain
planes. A detailed examination of all 48 point symmetries of the unstrained
diamond structure will be given in Section 3.5.2.
axes and under reflections through certain
planes. A detailed examination of all 48 point symmetries of the unstrained
diamond structure will be given in Section 3.5.2.
The point symmetries of the crystal structure are mirrored in the crystal potential, and hence in the one-particle Hamiltonian used for band structure calculations. Two important consequences for the electron band structure arise:
The irreducible wedge of the fcc lattice is depicted in Figure 3.5b. It has six corners
|
|
|
|
Previous: 3.3.3 Strain Resulting from Epitaxy Up: 3. Strained Bulk Band Structure Next: 3.4.1 Band Structure of Relaxed Si |