
 |
|
||||
BiographyLukas Cvitkovich was born in 1992 in Eisenstadt, Austria. He received his Bachelor degree in Electrical Engineering and his Diplom-Ingenieur degree in Material Sciences from TU Wien, where he defended his Master Thesis in the Quantum Materials group of the Institute of Solid State Physics in 2019. During his studies, he also worked as a research assistant at the Institute of Sensor and Actuator Systems and at the Institute for Solid State Electronics at TU Wien. In May 2020 he joined the Institute for Microelectronics where he started his PhD studies, focusing on the ab initio simulation of oxide defects in semiconductor devices. |
|||||
Computational Modeling of Initial Stage Si(100) Surface Oxidation
Semiconductor technology has relied on silicon and its native oxide SiO2 as a prime material system since the 1950s, due to its simple producibility and outstanding interface properties. In state-of-the-art device technology, materials with higher dielectric constants — so-called high-k materials — have replaced SiO2 as a gate dielectric. However, SiO2 is used to form passivation layers before the deposition of the high-k film, demanding fabrication of ultra-thin interfacial oxide layers. Typically, these layers are fabricated via thermal oxidation of Si, the physical mechanisms of which have been investigated for decades, both experimentally and theoretically. The classical standard model derived by Deal and Grove, while accurately describing the oxidation of Si in a well-progressed stage, strongly underestimates growth rates for thin oxide layers. On the other hand, fabrication of oxide layers with thicknesses in the nanometer range requires detailed knowledge of the oxidation mechanisms during initial oxidation. Furthermore, problems like the onset of amorphization or the interplay between various oxidation mechanisms are still unresolved. In order to address these problems, we investigated the initial stage of thermal oxidation of a Si(100) surface at T = 1000 K up to an oxide thickness of 2 nm by means of ab-initio molecular dynamics (AIMD) simulations in conjunction with Density Functional Theory (DFT), as well as the computationally cheaper Density Functional based Tight binding (DFTB) method. Within our simulations, we were able to reproduce all experimental observations that have been made on this subject until now. In addition, the data provides a qualitative explanation for the transition from the fast initial oxidation to the Deal-Grove oxidation process.
Our investigations start from the adsorption and dissociation of single O2 molecules. The enhanced growth rate at the onset of oxidation is explained by spontaneous surface reactions that are based on the transfer of an electron from the Si surface to the O2 molecule, triggering the dissociation of the molecule. The respective adsorption trajectory of each oxygen molecule strongly depends on its initial position and velocity. Qualitatively, the surface reactions are very similar, i.e., dissociative chemisorptions with energy gains of about 6 eV are observed for all adsorption events. The variety of adsorption trajectories indicate a stochastic oxidation process which leads to immediate amorphization of the oxide layer. A further increase of the oxygen coverage showed that the spontaneous surface reactions become less probable as the surface is oxidized. Instead, the O2 molecules adsorb to the surface into a metastable molecular precursor state that dissociates after not less than 3 ps. These precursor states block adsorption of oxygen molecules, and thus, effectively lower the rate of oxidation. Further supply of oxygen molecules resulted in an oxygen-saturated surface that consists of fully oxidized Si atoms, i.e., it forms a layer of amorphous SiO2 (see Fig. 1). Bond lengths and angles are in good agreement with experimental values of bulk a-SiO2. The saturation of the surface marks the beginning of the Deal-Grove oxidation regime that features slower oxygen incorporation via O2 diffusion through the oxide. As assumed within the Deal-Grove model, the O2 molecules dissociate at the Si/SiO2 interface. Our simulations show that the dissociation at the interface is again based on a charge transfer and happens spontaneously as soon as the molecule reaches the interface (see Fig. 2).
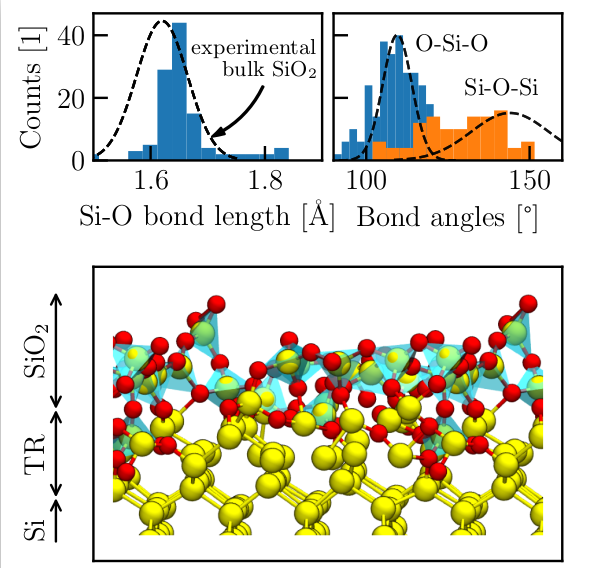
Fig. 1: Characteristics of the SiO2 growth obtained by dynamic simulations within DFTB. Distributions of the bond lengths and angles, and the comparison to experimental values of bulk SiO2 (dashed lines), are shown in the top panel. The lower panel shows the geometrically optimized structure after 32 O2 adsorptions. On Si atom (yellow) and four O atoms (red) form SiO4 tetrahedrons (blue surfaces) that cover the surface.
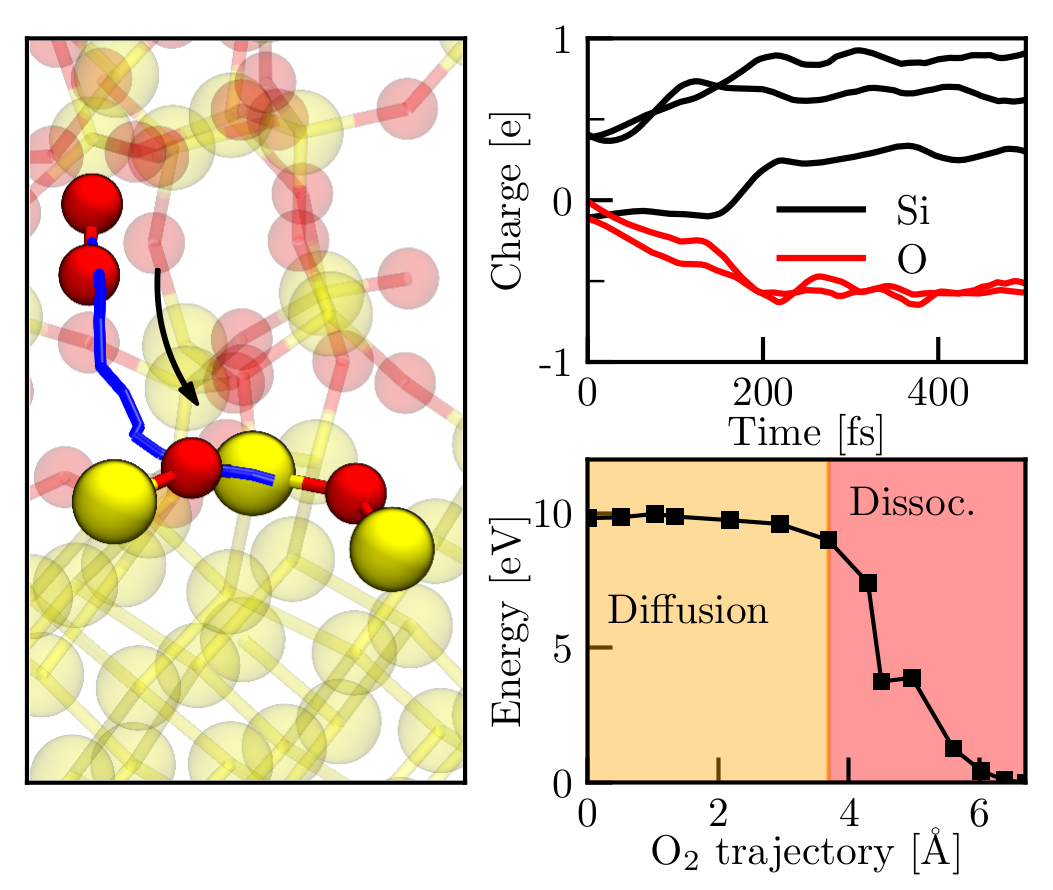
Fig. 2: O2 dissociation at the Si/SiO2 interface. The left panel shows the trajectory of the COM of the O2 molecule (solid blue line) together with the initial (molecular) and final (dissociated) configurations of the NEB calculation. The Mulliken charges for the spontaneous dissociation as captured within an AIMD simulation are depicted in the top right panel. The Mulliken analysis reveals another charge transfer of −e that strongly resembles the dissociative reactions at the surface during the initial stage of oxidation. The associated charges of two Si atoms show an offset due to the implications of the interface. The potential energy surface of this process is displayed in the lower right panel and indicates diffusion along a 4 Å long trajectory that is only inhibited by a low barrier of 0.2 eV. In addition to the AIMD results, the NEB calculation provides evidence for a spontaneous dissociation as soon as the molecule reached the dissociation spot.