
 |
|
||||
BiographyAnna Benzer obtained her her Master's degree in Computational Science and Engineering and Bachelor's in Technical Chemistry at the TU Wien. Anna joined the Institute for Microelectronics in November of 2023 as a doctoral student within the framework of the TU-D Doctoral College program "Unravelling Advanced 2D Materials (TU-D)". Her focus will be on using ab-initio and charge transport simulations to better understand the behavior of metal-doped 2D films as well as devices and gas sensors based on these. |
|||||
Density Functional Theory Study of Charged S Vacancies in MoS₂
Monolayer molybdenum disulfide (MoS2) is a promising material for next-generation electronic devices due to its atomically thin structure, mechanical flexibility, relatively high carrier mobility, and direct bandgap of approximately 1.8 eV. Its two-dimensional nature enables excellent electrostatic control and scalability, making it highly attractive for future sensors and digital logic devices.
To tailor the electronic and chemical properties of MoS2, substitutional doping with heteroatoms is a common strategy. However, a thorough understanding of intrinsic point defects, particularly sulfur vacancies, which are the dominant native defects, is essential before introducing extrinsic modifications. Sulfur vacancies significantly impact the material’s electronic structure and surface chemistry, influencing charge transport, adsorption/desorption kinetics of gas molecules, and contribute to hysteresis phenomena observed in electronic devices by acting as charge trapping centers.
A central goal is to model how MoS2 responds to exposure to various analyte gases (e.g., O2, N2), including charge trapping and hysteresis effects that can degrade device performance. For example, the localization of charge around an O2 molecule adsorbed at a sulfur vacancy is depicted in the figure below. Such phenomena are governed by defect charge transition levels (CTLs) and associated energy barriers, which can be investigated through Density Functional Theory (DFT) calculations combined with methods like the Nudged Elastic Band (NEB) technique. The accurate determination of CTLs requires calculating defect formation energies across different charge states. To address artifacts arising from long-range Coulomb interactions introduced by periodic boundary conditions, correction schemes must be applied to obtain reliable results for charged defects.
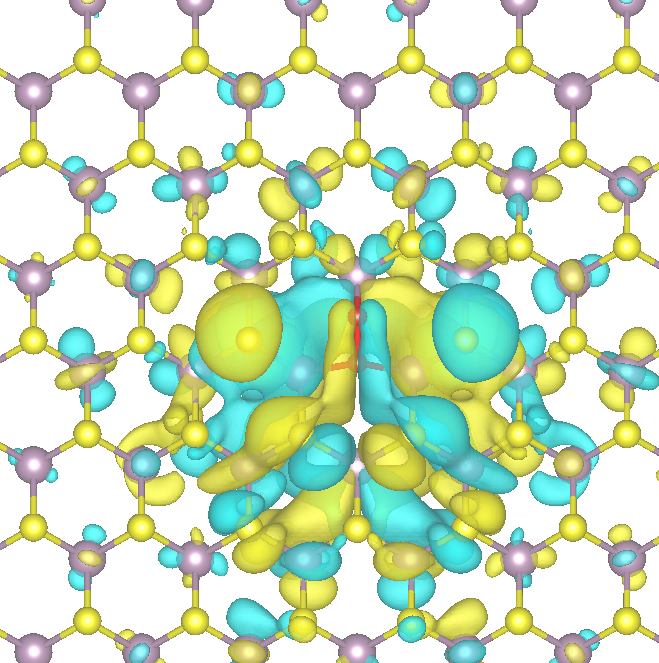
Fig. 1: Wave function visualized for O2 adsorbed in a S vacancy.