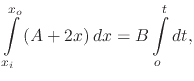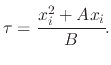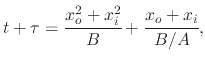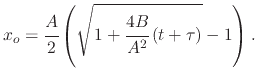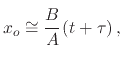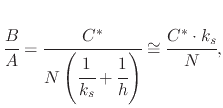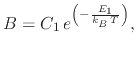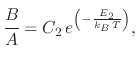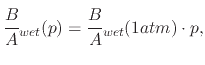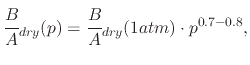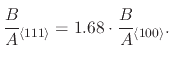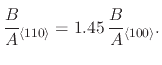Next: 2.3.2 Limitations of the Up: 2.3 Linear Parabolic Description Previous: 2.3 Linear Parabolic Description
The main idea behind the Deal-Grove model is illustrated in Figure 2.12 [42]. The figure represents the materials and interfaces involved during oxidation in a one-dimensional form. The transport and interaction of oxidants is viewed as going through the following stages:
The first step in the model is characterized by the surface reaction of free oxidants in the gas phase interacting with the oxide surface. The adsorption of oxidants through the top surface of the oxides is written as
Assuming an over-saturation of oxidant in the gas, ![]() is effectively the solubility limit in the oxide. This value is related
to the partial pressure in the atmosphere using Henry's law
is effectively the solubility limit in the oxide. This value is related
to the partial pressure in the atmosphere using Henry's law
The second flux ![]() from Figure 2.12 represents the diffusion of the oxidant from the oxide surface to the
oxide-silicon interface. Using Fick's law, the diffusion can be expressed as
from Figure 2.12 represents the diffusion of the oxidant from the oxide surface to the
oxide-silicon interface. Using Fick's law, the diffusion can be expressed as
The final flux ![]() presented in Figure 2.12 is the flux of oxidants consumed during the chemical reaction with
silicon atoms at the substrate surface, given by
presented in Figure 2.12 is the flux of oxidants consumed during the chemical reaction with
silicon atoms at the substrate surface, given by
Since steady-state conditions are assumed, the three fluxes representing the different stages of the oxidation process must be equal. The processes occur in series with each other and the rate of the overall process will be determined by the rate of the slowest process. Equating all fluxes results in
The overall oxidation rate is proportional to the flux of oxidant molecules,
The differential equation (2.15) can be simplified to
By integrating (2.15), from an initial oxide thickness ![]() to a final oxide thickness
to a final oxide thickness ![]() , a final result regarding
oxide kinetics can be written as
, a final result regarding
oxide kinetics can be written as
Introducing the simplified form from (2.16), (2.19) can be re-written to
Sometimes, it is useful to view (2.21) in the following form
A closer look at (2.23) suggests that there are two limiting forms of the linear parabolic growth law. The
parabolic or linear limiting form occur when
![]() or
or
![]() are the dominant terms
in (2.23), respectively. From (2.24), a limiting case can be identified when the oxidation time is given
by
are the dominant terms
in (2.23), respectively. From (2.24), a limiting case can be identified when the oxidation time is given
by ![]() and
and
![]()
The rate constants ![]() and
and ![]() which are the main idea behind the linear-parabolic oxide growth model are sometimes
referred to as the Deal-Grove parameters. These parameters have been extracted from experimental data and evaluated under a wide
range of experimental conditions [175].
which are the main idea behind the linear-parabolic oxide growth model are sometimes
referred to as the Deal-Grove parameters. These parameters have been extracted from experimental data and evaluated under a wide
range of experimental conditions [175].
The effects of temperature on the overall oxidation process have been examined in Section 2.2.1 where it was shown
that increasing the processing temperature resulted in an increased oxide thickness and a faster oxidation rate. Therefore, in
order to model oxidation using the linear-parabolic approach, both the linear (![]() ) and parabolic (
) and parabolic (![]() ) parameters
must be adjustable for temperature effects. From experimental data, it was found that Arrhenius expressions well describe
the temperature effects on
) parameters
must be adjustable for temperature effects. From experimental data, it was found that Arrhenius expressions well describe
the temperature effects on ![]() and
and ![]()
An analysis of the parabolic rate constant B from Table 2.2 shows that the activation energy ![]() for O
for O![]() and
H
and
H![]() O ambients are quite different. This suggests that the physical mechanism characterized by
O ambients are quite different. This suggests that the physical mechanism characterized by ![]() might be the
oxidant diffusion through SiO
might be the
oxidant diffusion through SiO![]() , since the diffusivity of O
, since the diffusivity of O![]() and H
and H![]() O in oxide are different,
O in oxide are different, ![]() is a constant value,
and
is a constant value,
and ![]() is not expected to exponentially increase with temperature. This suggests that the parameter
is not expected to exponentially increase with temperature. This suggests that the parameter ![]() from the linear parabolic
model represents the oxidant diffusion process.
from the linear parabolic
model represents the oxidant diffusion process.
The activation energy ![]() for
for ![]() in the table seems to be close to 2
in the table seems to be close to 2![]() for a O
for a O![]() system as well as a H
system as well as a H![]() O system.
This suggests that the physical origin of
O system.
This suggests that the physical origin of ![]() might be the chemical reaction at the silicon-silicon dioxide interface
might be the chemical reaction at the silicon-silicon dioxide interface ![]() .
The 2
.
The 2![]() activation energy has been associated with the Si-Si bond breaking process as confirmed by measurements performed by
Pauling, which suggested the correlation between the
activation energy has been associated with the Si-Si bond breaking process as confirmed by measurements performed by
Pauling, which suggested the correlation between the
![]() values and the activation energies of Si-Si bond
breaking [170].
values and the activation energies of Si-Si bond
breaking [170].
The effects of pressure on the oxide growth kinetics have been examined in Section 2.2.1, where it was shown that
increasing pressure causes an increased oxide film thickness when temperature is kept constant. Henry's law, relating to oxide
growth shown in (2.11) suggests a linear relationship between pressure and the oxidation rate. Since ![]() is proportional
to
is proportional
to ![]() , from (2.11) and both
, from (2.11) and both ![]() and
and ![]() are proportional to
are proportional to ![]() from (2.18)
and (2.27), respectively, then the growth rate should be proportional to
from (2.18)
and (2.27), respectively, then the growth rate should be proportional to ![]() .
Experimental measurements of H
.
Experimental measurements of H![]() O oxidation have shown this prediction to be correct for pressures ranging from
below to well above atmospheric [175]:
O oxidation have shown this prediction to be correct for pressures ranging from
below to well above atmospheric [175]:
However, in the case of dry oxidation with O![]() , the situation is somewhat unclear. Experimental results have consistently
shown that a linear relationship does not exist between the linear and parabolic rate constants and the applied pressure.
In fact, the linear rate constant is proportional to the pressure
, the situation is somewhat unclear. Experimental results have consistently
shown that a linear relationship does not exist between the linear and parabolic rate constants and the applied pressure.
In fact, the linear rate constant is proportional to the pressure
![]() , but the parabolic rate constant
varies with
, but the parabolic rate constant
varies with
![]() , where
, where ![]() . Since the linear rate constant is proportional to pressure, it
can be concluded that (2.11) is correct and
. Since the linear rate constant is proportional to pressure, it
can be concluded that (2.11) is correct and
![]() , but that the rate of reaction at the silicon
surface
, but that the rate of reaction at the silicon
surface ![]() depends on
depends on ![]() in a nonlinear fashion. In order to adjust the Deal-Grove model to satisfy the pressure effects in
dry oxidation, the values of
in a nonlinear fashion. In order to adjust the Deal-Grove model to satisfy the pressure effects in
dry oxidation, the values of ![]() and
and ![]() should be modified by:
should be modified by:
The crystal orientation of the oxidized silicon surface affects the oxide growth kinetics, as examined in Section 2.2.1. This effect has been observed even before the Deal-Grove model was suggested [126]. In order to associate the differences in oxidation kinetics with varying silicon crystal orientation, an analysis regarding the linear and parabolic rate constants in needed.
When observing the linear rate constant, except at the initial stage of oxidation, the oxide grows on silicon in an
amorphous way. Therefore, no information regarding the crystal structure of the underlying silicon is known as the oxide
volume increases. The linear rate constant ![]() should not change with a changing crystal orientation of the underlying silicon.
This is also observed in experiments by extracting growth data for various crystal orientations [122].
should not change with a changing crystal orientation of the underlying silicon.
This is also observed in experiments by extracting growth data for various crystal orientations [122].
However, the parabolic rate constant ![]() should depend on the silicon crystal orientation. The reason is that it
involves the chemical reaction which occurs directly on the Si/SiO
should depend on the silicon crystal orientation. The reason is that it
involves the chemical reaction which occurs directly on the Si/SiO![]() interface. The speed of this reaction should depend on
the amount of silicon atoms available for the reaction. It was found experimentally that surfaces which provide more available
reaction sites to silicon have a higher oxidation rate [122]. The ratio for the parabolic rate constant in
silicon crystal orientations (111):(100) was found to be 1.68:1. This can be adjusted in the Deal-Grove model by
interface. The speed of this reaction should depend on
the amount of silicon atoms available for the reaction. It was found experimentally that surfaces which provide more available
reaction sites to silicon have a higher oxidation rate [122]. The ratio for the parabolic rate constant in
silicon crystal orientations (111):(100) was found to be 1.68:1. This can be adjusted in the Deal-Grove model by
![\includegraphics[width=0.70\linewidth]{chapter_oxidation/figures/one-dimDG.eps}](img176.png)
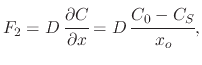
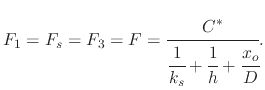
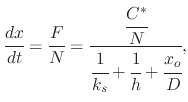
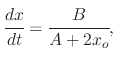
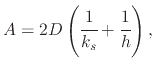
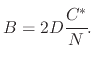
![$\displaystyle N\intop_{x_{i}}^{x_{o}}\left[1+\cfrac{k_{s}}{h}+\cfrac{k_{s}x}{D}\right]dx=k_{s}C^{*}\intop_{0}^{t}dt.$](img201.png)