
 |
|
||||
BiographyLukas Cvitkovich was born in 1992 in Eisenstadt, Austria. He received his Bachelor degree in Electrical Engineering and his Diplom-Ingenieur degree in Material Sciences from TU Wien, where he defended his Master Thesis in the Quantum Materials group of the Institute of Solid State Physics in 2019. During his studies, he also worked as a research assistant at the Institute of Sensor and Actuator Systems and at the Institute for Solid State Electronics at TU Wien. In May 2020 he joined the Institute for Microelectronics where he started his PhD studies, focusing on the ab initio simulation of oxide defects in semiconductor devices. |
|||||
Multiscale Modelling of the Formation of Amorphous SiO2
Silicon and oxygen are without a doubt the most important elements in present-day metal-oxide semiconductor devices. Not only is the Si/SiO2 interface unrivaled in quality and usability for semiconductor technology, but the interplay between these two elements gains importance since oxygen is also the most common impurity in commercially grown single-crystalline silicon. With increasing demand for smaller devices, understanding the initial oxidation process, as well as the atomic structure of Si/SiO2 interfaces, their growth and the migration of oxygen into the silicon substrate, is becoming more important and has raised interest in large-scale atomic simulations that can help to shed light on these issues.
The oxidation process of crystalline Si forming amorphous SiO2 was studied using multiple atomic computer simulation techniques for various system sizes. Starting with small systems consisting of 226 atoms, the adsorption and dissociation of single O2 molecules onto an Si[100] surface was investigated by means of ab-initio molecular dynamics (AIMD).
These simulations show that the dissociative oxygen adsorption onto the Si surface is facilitated by a charge transfer from the reconstructed Si dimers at the surface (Fig. 1). Due to the high computational costs of AIMD, the oxidation process of multiple O2 molecules was examined using the less expensive Density Functional-Based Tight Binding method (DFTB) on larger systems containing up to 900 atoms. The actual formation of SiO2 was modeled within molecular dynamics simulations (MD) utilizing reactive force-fields (ReaxFF) and a modified/parametrized Stillinger-Weber potential resulting in atomic SiO2 structures with sizes of about 5000 atoms (Fig. 2). The barriers for oxygen diffusion between bond-center sites in a silicon bulk have been calculated using the NEB technique. The results suggest that the migration of O atoms in Si is prevented at reasonable temperatures, which is in accordance with the MD simulations. Instead, evidence for a strain-driven diffusion mechanism has been found on various scales, resulting in an oxidation front that represents a Si/SiO2 interface.
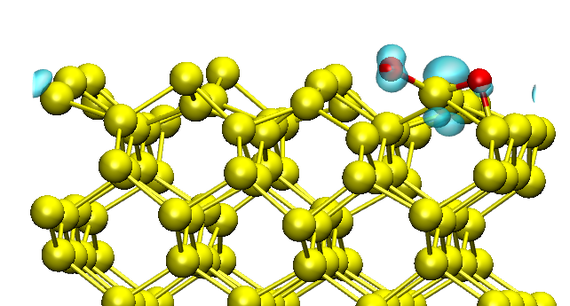
Fig. 1: Spin-restricted ab-initio molecular dynamics simulation of the dissociative chemisorption of a triplet oxygen (red) to the Si surface. The isosurface of the spin density is shown in blue.
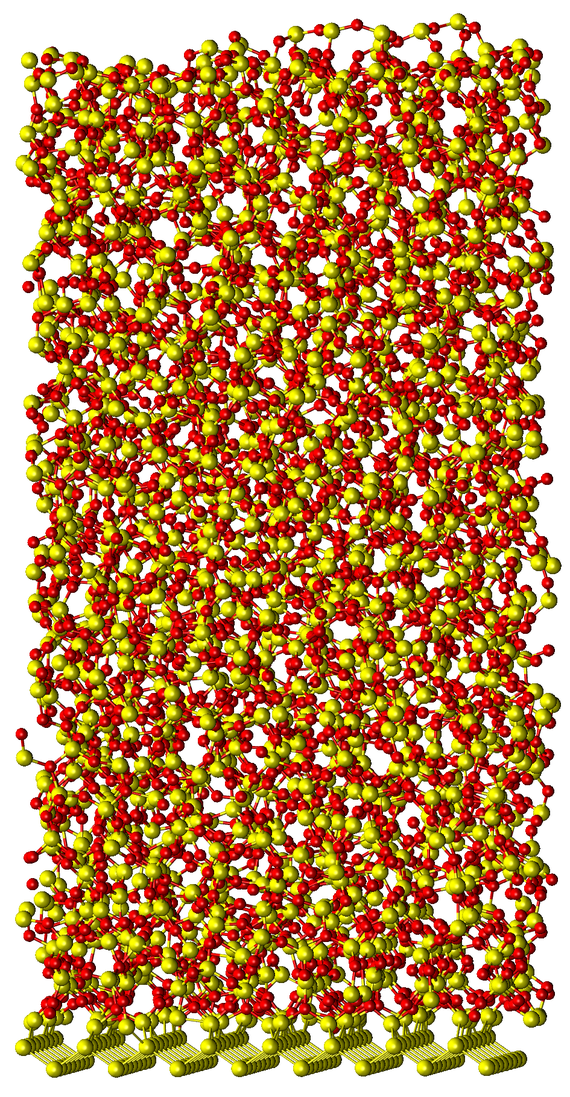
Fig. 2: SiO2 structure growth on a Si substrate using molecular dynamics simulations.