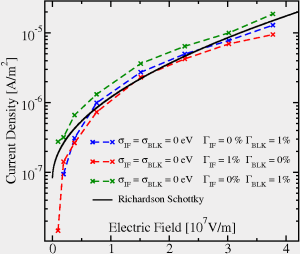
|
A computer program for the three-dimensional microscopic simulation of transient phenomena in amorphous organic devices has been developed. The device physics of polymer-based opto-electronic applications is characterized by the injection, propagation, extraction and accumulation of charges in layered assemblies of about 100 nm thin polymer films at field strengths of about 1 MV/cm. Electrons in amorphous plastic devices behave noticeably differently than conduction electrons within a periodically ordered crystal. Whereas the latter delocalize across the translation invariant bulk, electrons in semiconducting plastics equal point charges on an erratic journey through the polymer. The resulting electric currents are often randomly distributed in space and frequently give rise to mobility models based on the percolation theory. The software presented is based on the Kinetic Monte Carlo technique and thus simulates the system's time evolution as a continuous time random walk. The simulator drives both the time-evolution of the p- and the n-conductive band simultaneously. Whether the device is n- or p-conductive does not depend on an intrinsic material property, but on the constellation of work functions. All space charge and image-force effects have been regarded with full rigor. Electrical doping can be simulated on an arbitrary spatial doping level using arbitrary spatial dopant distributions. Disorder is modeled by Baessler's well-established Gaussian Disorder Model. The transition of electrons is governed by the classical Miller-Abrahams expression, which turned out to be adequate for bulk and hetero-junction simulations, as countless Monte Carlo simulations by Baessler and many other researchers in this area prove. However, many organic devices exhibit contact-dominated behavior. In this case, the bulk's molecular structure is less critical to a device's electrical characteristics than the chemical and morphologic structure of the bulk-electrode interfaces. Since the Miller-Abrahams rate is inadequate for the simulation of charge injection and extraction in real space, it has currently been replaced by an advanced mechanism.
|