

 Previous: 6. Conclusion and Outlook
Up: Dissertation A. Hoessinger
Next: Bibliography
Previous: 6. Conclusion and Outlook
Up: Dissertation A. Hoessinger
Next: Bibliography
The list of command-line parameters of MCIMPL is available by executing the
simulator with the command-line option -help.
MCIMPL -help
The simulation is started by executing
MCIMPL inputName outputName [-option Value]
The required parameters are:
| inputName |
Name of the file which contains the topological information of the
simulation domain in binary PIF-Format ([22] [21]). |
| |
|
| outputName |
Name of the output file. It is a file in binary PIF-Format which
contains the distribution of the implanted particles and the distribution of the
material damage. If a three-dimensional simulation is performed the
distributions are based on an ortho-grid, in case of a two-dimensional
simulation a geometry conforming triangular grid is generated. The output file
of a two-dimensional simulation additionally contains the topological
information provided on input. |
As well the name of the input file as of the output file are strings which may
also contain path information according to Unix file system specifications.
In the following all options of the simulator are listed, since all of them are
optional the options describing the implantation conditions should be set by the
user. The default values for the implantation conditions should just be used for
testing the simulator. The options describing the implantation conditions
are:
|
Name of the implanted ion species. This name must be contained in the model
parameter file mcimpl_data.dat (Sec. 2.2.1). The value of this
option is a string. The default value is boron. |
| |
|
|
The energy of the implanted ions in keV (Sec. 2.2.2). The value of this
option is a single floating point number. The default value is 25. |
| |
|
|
The implantation dose in atoms/cm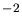 (Sec. 2.2.3).The value of this
option is a single floating point number. The default value is (Sec. 2.2.3).The value of this
option is a single floating point number. The default value is
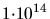 . . |
| |
|
|
The angle between the ion beam and the wafer normal
(Sec. 2.2.4). The value of this option is a single floating
point number. The default value is 0. |
| |
|
|
The rotation angle of the ion beam related to the primary flat of the wafer
(Sec. 2.2.4). The value of this option is a single floating
point number. The default value is 0. |
| |
|
|
Number of ions used for the simulation. Only the number of ions
started from outside of the simulation domain is set by this option. The number
of ion trajectories that are generated through the simulation is determined by
other parameters or by the applied models (Follow
Each Recoil model). The higher the number of simulated ions the higher the
accuracy of the simulation results. This number is just an approximate
value. The real number of simulated ions is additionally influenced by the size
of the simulation domain and the implantation conditions. The value of this
option is a single integer number. The default value is 200000. For one-dimensional applications a value of the order of 10000 should
be used. For two-dimensional applications about 100000 ions per 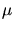 m should
be used and for three-dimensional applications 2000000 ions per m m should
be used and for three-dimensional applications 2000000 ions per m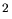 are
recommended. are
recommended. |
Some special process parameters can be defined if necessary:
|
One simulation can be performed with several rotation angles
(Sec. 4.5.4). This option specifies the number of different rotation
angles. The value of this option is a single integer number. The
default value is 0, which means that just one ion beam direction is used
during the implantation. A value of one equals two ion beam directions with a
difference in the rotation angel of 180
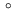 . . |
| |
|
|
The wafer is rotated continuously during the
simulation. It is a binary option which has no value. By specifying
-revolving this option is activated, by specifying -norevolving this
option is deactivated. By default this option is deactivated. |
| |
|
|
Divergence of the ion beam in
(Sec. 4.3). The value of this
option is a single floating point number. The default value is 0. |
| |
|
| -crystalOrientation |
| -crystal |
|
This option determines the orientation of the substrate crystal, by defining
the miller indices of the crystal direction which is parallel to the
z-axis. The x-axis of the simulation domain is always parallel to the 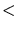 100 100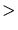 direction. The value of this option is a string. Only ``100'' or
``110'' or ``111'' are allowed values. The default value is ``100''.
direction. The value of this option is a string. Only ``100'' or
``110'' or ``111'' are allowed values. The default value is ``100''. |
| |
|
|
This option determines the wafer temperature, but it has no influence on the
simulation behavior, because the lattice vibration is not derived from this
temperature but from the option -atomVibration. This
option is just used to calculate the critical damage energy which is an output
value of the simulator. The value of this option is a single floating
point number. The default value is 293. |
| |
|
|
The average amplitude of the thermal lattice vibration in units of the
lattice constant (Sec. 3.3.4). The value of this option is a single floating point number. The default value is 0.009. |
The following options are used to specify the input and the output of the
simulator:
|
Each physical PIF file may contain several logical files. This option
determines the name of the logical file. The value of this option is a string. The default value is MC_In. If the value dose not coincide
with any logical file in the physical PIF file the first logical file in the
physical PIF file is selected. If the physical PIF file contains just one
logical file this option needs not to be defined. |
| |
|
|
The output PIF file contains one logical file. This option sets the name of
this logical file. The value of this option is a string. The default
value is MC_Out. |
| |
|
|
Specifies whether the damage information generated by a preceding MCIMPL
simulation run shall be reused (Sec. 4.5.1). It is a binary option. By default it is deactivated. |
| |
|
|
A point response function is calculated which can
be used by the analytical ion implantation simulator IMP3D (Sec. 3.2,
Sec. 4.5.3). It is a binary option. By default it is deactivated. |
| |
|
|
Determines that one file in ASCII format is generated for each
impurity species which only represents the z-dependence of the impurity
distribution. In case of a one-dimensional simulation this equals the simulation
result. In case of higher dimensional simulations it is an average over the
other coordinates in space. It is a binary option. By default it is deactivated. |
| |
|
| -backupIntervall |
| -backInt |
|
Determines that a backup file of the current simulation status is
generated regularly after a certain amount of ions has been simulated. The name
of the backup file equals the name of the output file without the extension
.pbf, but with the extension _BCK.DAT. The value of
this option is a single integer number. The default value is 50000. |
| |
|
|
The backup file is used to restart a simulation. It is a binary
option. By default it is deactivated. |
| -implantationWindow |
| -implWin |
|
If not the whole surface of the simulation domain shall be exposed to the ion
implantation simulation a rectangular implantation window can be specified with
this option. In case if a point response function is calculated this window has
to be determined to identify the position of the significant point and to define
the size of the area which is averaged (Sec. 4.2). The value of this option
is a list of six floating point numbers, which determine the front
left most point (first three values) and the back right most point of the window
(last three values) (x1 y1 z1 x2 y2 z2). By default an automatic implantation
window is created. |
| |
|
|
In case of a two-dimensional simulation a triangular grid is generated. This
options specifies that all points of the grids contained in the input file
should be reused for the generation of the grids for the output file. It is a
binary option. By default it is deactivated. |
| |
|
|
Minimum acceptable angle in a triangular grid element (only relevant for
two-dimensional simulations). The value of this option is a single floating point
number and the unit is
. The default value is 20.0
. |
| |
|
|
Maximum acceptable relative dose error in a triangular grid element (only
relevant for two-dimensional simulations). The value of this option is a single
floating point number. The default value is 0.1. |
| |
|
|
Maximum acceptable concentration (logarithmic) gradient in a triangular grid
element (only relevant for two-dimensional simulations). The value of this option is a single floating point number. The default value is 3.0. |
| |
|
| -[no]smoothOutput |
| -smooth |
|
A smoothing operation is performed on the simulation results. It is a binary option. By default it is activated. |
Several parameters are used to choose between models and to determine additional
conditions for models:
|
Determines if material damage is considered during the
simulation. It is a binary option. By default it is activated. |
| |
|
|
Defines the recoil species that are handled by the Follow-Each-Recoil
method. By default silicon and oxygen are considered
(Sec. 3.3.5). The value of this option is a list of
stings. Each string determined the name of one atom species in small
letters. |
| |
|
| -[no]simpleMolecule |
| -smpl |
|
If simpleMolecule is activated molecular ions are treated as one big ion with a
modified damage generation behavior. Just the distribution of one
(significant) atom species is calculated (Sec. 4.5.2). It is a binary option. By default it is activated. |
| |
|
|
Specifies the approximate number of initial ions. For the initial
ions no Trajectory-Split method is applied, even if the Trajectory-Split method is
enabled. The value of this option is a single integer number. The
default value is 0. Care has to be taken that the value is not too high,
because this can influence the simulation result. The number of primary ions
should not be larger than one hundredth of the total number of simulated ions. |
| |
|
|
The type of Trajectory-Split method which is applied ([9]). The
value of this option is a string. Only ``TRS'' and ``SRS'' are
allowed values. The default value is TRS. |
| |
|
|
This option does not influence the behavior of the simulation. The value
specified is only written to the point response file for the analytical
simulator (only relevant for point response calculation). The analytical
simulator can use this parameter as an additional parameter to characterize
point response functions. The value of this option is a single floating
point number. The default value is 0.0. |
| -[no]kinchinPeaseModel |
| -kinch |
|
Determines the damage model used for the simulation. Either the
Kinchin-Pease model or the Follow-Each-Recoil model is used. This option is only
effective if the use of material damage is enabled. It is a binary
option. By default it is activated. |
| |
|
| -simulationDepth |
| -simDepth |
|
In case of a three-dimensional simulation the simulation domain is
discretized by an octree. The depth of the root cube in (
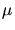 m) can be set by this
option. The value of this option is a single floating point
number. The default value is the depth of the simulation domain. Care has to be
taken that this value is not smaller than the penetration depth of the ions into
the simulation domain. If this is the case some ions get lost. m) can be set by this
option. The value of this option is a single floating point
number. The default value is the depth of the simulation domain. Care has to be
taken that this value is not smaller than the penetration depth of the ions into
the simulation domain. If this is the case some ions get lost. |
| |
|
|
Defines the resolution of the geometry discretization by the
octree (only relevant for three-dimensional simulations) [79]
[78]. The value of this option is a single integer number,
which may be in the interval from 1 to 10. The default value is 9. |
| |
|
|
Determines how often one trajectory may be split
(Sec. 4.6.1). The value of this option is a single integer number,
which may be in the interval from 0 to 10. The default value is 8. |
Finally there are some options which set parameters used by physical or empiric
models, and which my significantly influence the behavior of the
simulator. This options should only be set by expert users when developing new
models or when calibrating existing models:
|
If the particle energy falls below this limit, the trajectory calculation is
stopped. The energy unit is eV. The value of this option is a single
floating point number. The default value is 10.0. |
| |
|
| -displacementEnergy |
| -displEng |
|
The energy which is necessary to remove an atom of the target from its
lattice position. This displacement energy is applied to all materials and atom
species in the simulation domain. The energy unit is eV. The value of this
option is a single floating point number. The default value is 15.0. |
| -screeningPrefactor |
| -screenPre |
|
Parameter in the empirical electronic stopping model
(Sec. 3.3.3). The value of this option is a list of
floating point numbers. One value has to be specified for each atom species
in the implanted molecule. In case of single atomic ion implantations this list
has only one element. The species are ordered according to the order
in the chemical formula of the molecule. For instance in case of BF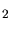 ion
implantation two floating point numbers have to be defined. The first one is
applied to the boron atom and the second one to the fluorine atom. The default values
are taken from the file mcimpl_data.dat. ion
implantation two floating point numbers have to be defined. The first one is
applied to the boron atom and the second one to the fluorine atom. The default values
are taken from the file mcimpl_data.dat. |
| |
|
| -lindhardCorrection |
| -lindCorr |
|
Parameter in the empirical electronic stopping model
(Sec. 3.3.3). The value of this option is a list of
floating point numbers. One value has to be specified for each atom species
in the implanted molecule. In case of single atomic ion implantations this list
has only one element. The species are ordered according to the order
in the chemical formula of the molecule. For instance in case of BF ion
implantation two floating point numbers have to be defined. The first one is
applied to the boron atom and the second one to the fluorine atom. The default values
are taken from the file mcimpl_data.dat. |
| |
|
| -nonlocalPrefactor |
| -nlPre |
|
Parameter in the empirical electronic stopping model
(Sec. 3.3.3). The value of this option is a list of
floating point numbers. One value has to be specified for each atom species
in the implanted molecule. In case of single atomic ion implantations this list
has only one element. The species are ordered according to the order
in the chemical formula of the molecule. For instance in case of BF ion
implantation two floating point numbers have to be defined. The first one is
applied to the boron atom and the second one to the fluorine atom. The default values
are taken from the file mcimpl_data.dat. |
| |
|
|
Parameter in the empirical electronic stopping model
(Sec. 3.3.3). The value of this option is a list of
floating point numbers. One value has to be specified for each atom species
in the implanted molecule. In case of single atomic ion implantations this list
has only one element. The species are ordered according to the order
in the chemical formula of the molecule. For instance in case of BF ion
implantation two floating point numbers have to be defined. The first one is
applied to the boron atom and the second one to the fluorine atom. The default values
are taken from the file mcimpl_data.dat. |
| -recombinationFactor |
| -recFac |
|
Parameter in the empirical damage recombination model used in common with the
Kinchin-Pease model (Sec. 3.3.5). The value of this option is a list of
floating point numbers. One value has to be specified for each atom species
in the implanted molecule. In case of single atomic ion implantations this list
has only one element. The species are ordered according to the order
in the chemical formula of the molecule. For instance in case of BF ion
implantation two floating point numbers have to be defined. The first one is
applied to the boron atom and the second one to the fluorine atom. The default values
are taken from the file mcimpl_data.dat. |
| |
|
|
Parameter in the empirical damage recombination model used in common with the
Kinchin-Pease model (Sec. 3.3.5). The value of this option is a list of
floating point numbers. One value has to be specified for each atom species
in the implanted molecule. In case of single atomic ion implantations this list
has only one element. The species are ordered according to the order
in the chemical formula of the molecule. For instance in case of BF ion
implantation two floating point numbers have to be defined. The first one is
applied to the boron atom and the second one to the fluorine atom. The default values
are taken from the file mcimpl_data.dat. |
| |
|
|
Parameter in the recombination model used in combination with the Follow Each
Recoil method (Sec. 3.3.5). The value of this option is a single
floating point number. The unit of this option is the silicon lattice
constant. The default value is 1.0. |
| |
|
| -nbConcentrationLevel |
| -nbConc |
|
The Trajectory-Split method uses concentration levels to decide whether an ion
is split or not. This options sets the number of these concentration
levels. Increasing this number increases the probability of splitting
(Sec. 4.6.1). The value of this option is a single
integer number in the interval from 1 to 10. The default value is 10. |
| |
|
| -relativeSplitEnergy |
| -relSpEnerg |
|
Sets an energy threshold for the Trajectory-Split method. If the
ion energy is above this value no splitting is allowed. It is a relative value
to the initial ion energy (Sec. 4.6.1). The value of this option is a
single floating point number. The default value is calculated by an
empirical formula. |
| -relativeSplitLength |
| -relSpLen |
|
The length of the ion trajectory must exceed this value before splitting is
allowed. It is relative value to the average trajectory length
(Sec. 4.6.1). The value of this option is a single floating
point number. The default value is calculated by an empirical formula. |
| |
|
| -searchDistance |
| -searchDis |
|
Parameter which specifies the distance from the ion position, for looking for
potential collision partners used by the nuclear and electronic stopping models
(Sec. 4.4.1). The value of this option is a single floating
point number. The unit is the silicon lattice constant and the default value is
3.0. |
| |
|
| -minScatteringAngle |
| -minScat |
|
This parameter is used when selecting collision partners. If a nuclear
scattering event results in a scattering angle above the value specified, the
check for collision partners involved in the preceding collision process is
omitted when initializing the next stopping process (Sec. 4.4.1). The value
of this option is a single floating point number. The unit is degree
and the default value is 30.0. |
| |
|
| -minFlightPath |
| -minFlight |
|
If the flight path of a stopping process falls below the value specified the
list of lattice sites involved in the stopping process is not replaced by a new
list, but the list of the previous stopping process is extended. This list is
used to avoid multiple collisions with the same target atoms
(Sec. 4.4.1). The value of this option is a single floating point
number. The unit is the silicon lattice constant and the default value is 0.05. |
Previous: 6. Conclusion and Outlook
Up: Dissertation A. Hoessinger
Next: Bibliography
A. Hoessiger: Simulation of Ion Implantation for ULSI Technology