Next: 2.5.1 Erste Born-Näherung Up: 2 Theorie der Störstellenstreuung Previous: 2.4 Born-Wirkungsquerschnitt erster Ordnung
![]()
![]()
![]()
Next: 2.5.1
Erste Born-Näherung Up: 2
Theorie der Störstellenstreuung Previous: 2.4
Born-Wirkungsquerschnitt erster Ordnung
Der im letzten Abschnitt hergeleitete Ausdruck
(2.24) für das Streupotential zweier
benachbarter, räumlich ausgedehnter Störstellen in einem dotiertem
Halbleiter ist exakt, wenn man von Mehrfachstreuungen absieht. Um jedoch
den differentiellen Wirkungsquerschnitt (2.30)
der ionisierten Störstellenstreuung von Elektronen an einem bestimmten
Dopanden berechnen zu können, benötigen wir neben der räumlichen
Verteilung der Störstellen auch den Formfaktor des entsprechenden
Dopanden, also die räumliche Ladungsverteilung seiner Valenz- und
Kernelektronen. Da wir an analytischen Ausdrücken interessiert sind,
bedienen wir uns des klassischen Atommodells von Thomas [Tho27]
und Fermi [Fer27] zur Berechnung der Ladungsdichte,
wobei wir jedoch die Ladungsdichte durch Variation des totalen Energiefunktionals
und nicht über die TF Gleichung berechnen wollen. Das ursprüngliche
TF Modell hat seit damals viele Verbesserungen erfahren [GSD71],
sodaß man schließlich durch Hinzunahme einer Austauschkorrektur
nach Dirac [Dir30] und einer Gradientenkorrektur![]() zur kinetischen Energie nach Weizsäcker [vW35]
eine bemerkenswert erfolgreiche (TFDW) Theorie zur Verfügung hat,
die für große Ordnungszahlen exakt mit quantenmechanischen Modellen
übereinstimmt [Thi90]. Das TF Atommodell
geht von einem entarteten Fermigas bei Null Kelvin aus, das in einem konstant
positiven Untergrund eingebettet ist [Gom49,Sca96].
Die Ladung der Valenz- und Kernelektronen wird also kontinuierlich verteilt
angenommen und kann als eine Atmosphäre um die Kerne betrachtet werden.
Durch die vollständige Entartung erhält man eine lokale Beziehung
zwischen der Elektronendichte und der Fermienergie. Dieses statistische
Atommodell des entarteten Elektronengases liefert etwa überraschenderweise
genaue Ergebnisse für die Ionisationsenergien verschiedener Atome
[KG73], kann jedoch Phänomene, die auf
individuellen Eigenschaften einzelner Elektronen beruhen, naturgemäß
nicht erklären (Periodensystem). Die hohe Entartung der Valenz- und
Kernelektronen dürfte der Grund dafür sein, daß die Näherung
des Elektronensystems durch ein entartetes Fermi-Gas bei Null Kelvin überraschend
gut ist [FST86].
zur kinetischen Energie nach Weizsäcker [vW35]
eine bemerkenswert erfolgreiche (TFDW) Theorie zur Verfügung hat,
die für große Ordnungszahlen exakt mit quantenmechanischen Modellen
übereinstimmt [Thi90]. Das TF Atommodell
geht von einem entarteten Fermigas bei Null Kelvin aus, das in einem konstant
positiven Untergrund eingebettet ist [Gom49,Sca96].
Die Ladung der Valenz- und Kernelektronen wird also kontinuierlich verteilt
angenommen und kann als eine Atmosphäre um die Kerne betrachtet werden.
Durch die vollständige Entartung erhält man eine lokale Beziehung
zwischen der Elektronendichte und der Fermienergie. Dieses statistische
Atommodell des entarteten Elektronengases liefert etwa überraschenderweise
genaue Ergebnisse für die Ionisationsenergien verschiedener Atome
[KG73], kann jedoch Phänomene, die auf
individuellen Eigenschaften einzelner Elektronen beruhen, naturgemäß
nicht erklären (Periodensystem). Die hohe Entartung der Valenz- und
Kernelektronen dürfte der Grund dafür sein, daß die Näherung
des Elektronensystems durch ein entartetes Fermi-Gas bei Null Kelvin überraschend
gut ist [FST86].
Das Grundproblem bildet nun die Bestimmung der Potentialverteilung der Elektronen eines atomaren Systems. Die Lösung dieses Problems fanden unabhängig voneinander unter den obigen Annahmen Thomas [Tho27] und Fermi [Fer27]. Da die TF-Gleichung nur numerisch lösbar ist, bedienen wir uns des Ritz'schen Variationsverfahrens zur Bestimmung der Ladungsdichte. Genauso wie man die Schrödinger-Gleichung aus einem Variationsprinzip herleiten kann, läßt sich auch die TF-Gleichung aus einem Variationsansatz herleiten, indem man das totale Energiefunktional bildet [Len32]. Man gibt eine beliebige Ladungsverteilung mit zunächst unbestimmten Parametern vor und berechnet die totale Energie. Die Parameter werden dadurch bestimmt, daß die Energie ein Minimum annehmen muß. Je besser die gewählte Ladungsdichte der echten entspricht, desto genauer ist der erhaltene Wert für die minimale Energie, die der experimentell gemessenenen Ionisierungsenergie im Grundzustand entspricht. Der Variationsansatz ist der herkömmlichen TF-Methode, die sich der TF-Gleichung bedient, überlegen, da man durch entsprechende Wahl von Funktionen der quantenmechanischen Lösung sehr nahe kommt.
Die totale Energie E des Systems setzt sich zusammen aus der
klassischen elektrostatischen Energie ![]() aufgrund der Elektron-Elektron-Wechselwirkung, der Coulomb-Energie
aufgrund der Elektron-Elektron-Wechselwirkung, der Coulomb-Energie ![]() aufgrund der Elektron-Kern-Wechselwirkung, der kinetische Energie
aufgrund der Elektron-Kern-Wechselwirkung, der kinetische Energie ![]() ,
einer Inhomogenitätskorrektur
,
einer Inhomogenitätskorrektur ![]() für the kinetische Energie [vW35] und
einer quantenmechanischen Austauschkorrektur
für the kinetische Energie [vW35] und
einer quantenmechanischen Austauschkorrektur ![]() [Dir30]:
[Dir30]:
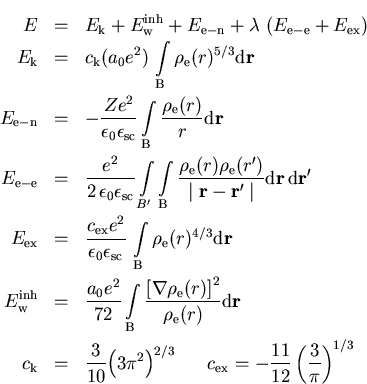 |
(2.31) |
wobei a0 der klassische Bohr-Radius ist (Anhang A).
Bei der Berechnung von ![]() wird vernachlässigt, daß ein Elektron nicht auf sich selbst
wirken kann. Der Korrelationsparameter
wird vernachlässigt, daß ein Elektron nicht auf sich selbst
wirken kann. Der Korrelationsparameter ![]() ,
der immer kleiner als eins ist, wird eingeführt, um diesen Fehler
auszugleichen. Durch Nullsetzen der partiellen Ableitungen von
,
der immer kleiner als eins ist, wird eingeführt, um diesen Fehler
auszugleichen. Durch Nullsetzen der partiellen Ableitungen von ![]() nach den n Variationsparametern
nach den n Variationsparametern ![]()
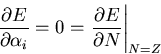 |
(2.32) |
erhält man ein Gleichungssystem vom Rang n+1, woraus man
die unbestimmten Parameter ![]() und
und ![]() in eindeutiger Weise erhält. Die letzte Gleichung spiegelt das Verschwinden
des chemischen Potentials eines neutrales Atom im TF Modell wider [Thi90].
in eindeutiger Weise erhält. Die letzte Gleichung spiegelt das Verschwinden
des chemischen Potentials eines neutrales Atom im TF Modell wider [Thi90].
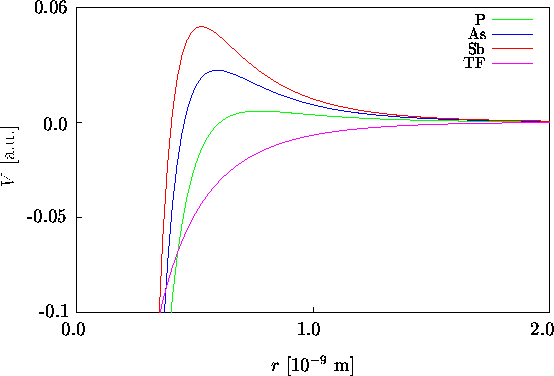 |
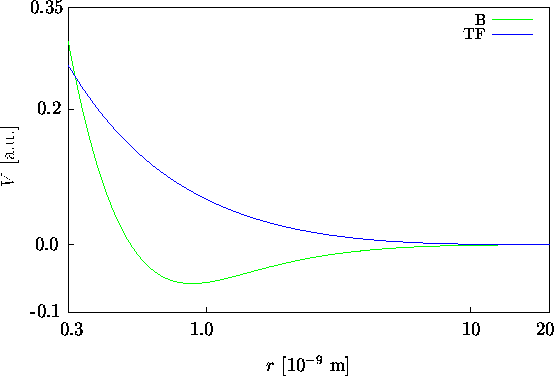
Wir wollen in weiterer Folge eine geschirmte Coulomb-Verteilung annehmen,
die die quantenmechanisch berechnete Ladungsdichte, die für große
r exponentiell abklingt, besser wiedergibt als die Lösung der
TF-Gleichung, die zu langsam abklingt. Die normierte, exponentiell abgeschirmte
Ladungsdichteverteilung und der entsprechende Formfaktor für N
Valenz- und Kernelektronen lautet
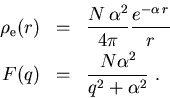 |
(2.33) |
Wenn wir nun (2.33) in (2.31)
einsetzen, erhalten wir für die totale Energie
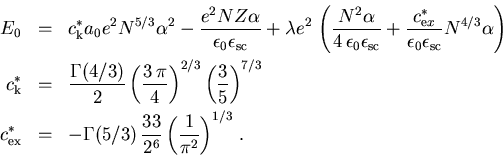 |
(2.34) |
Berechnungen haben interessanterweise gezeigt, daß die mit Hilfe
von (2.34) erhaltenen Grundzustandsenergien
nicht notwendigerweise besser sind als jene, die mit dem ursprünglichen
TF Energiefunktional berechnet werden, das keine quantenmechanischen Korrekturen
enthält [Sca96]. Nach Minimierung von
(2.34 erhalten wir für ![]()
![\begin{eqnarray}\alpha &=&\frac{Z^{1/3}}{c_{\mathrm k}^{*}a_{0}\epsilon_{0}\eps... ...3}} \right] }{\frac{5}{3}-4\,\left( \frac{Z}{N}\right)^{1/3}}\; .\end{eqnarray}](img365-2.gif) |
(2.35) |
Damit erhalten wir für den Korrelationsparameter
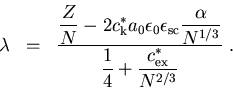 |
(2.36) |
![]() ist ein Maß für die mittlere räumliche Ausdehnung der Ladungsverteilung
der Valenz- und Kernelektronen der Störstelle.
ist ein Maß für die mittlere räumliche Ausdehnung der Ladungsverteilung
der Valenz- und Kernelektronen der Störstelle.
 |