« PreviousUpNext »Contents
Previous: 3.5 Void Nucleation Top: 3 Models Next: 4 Numerical Implementation
3.6 Void Evolution
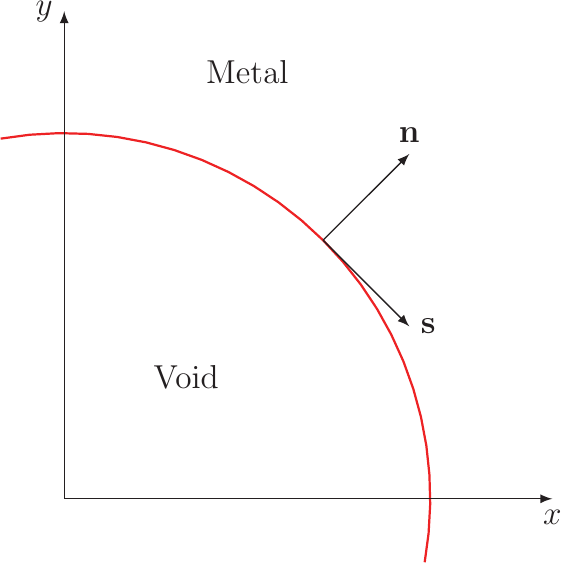
The void evolution phenomenon causes degradation by increasing the resistance of an interconnect after the void has nucleated. This process is modeled by equations tracking the movement of an interface, representing the boundary between the interconnect metal and the void [73]. For the calculations Bhate et al. [8, 9] introduced a local coordinate system at the void surface (shown in Figure 3.4). The chemical potential of the surface, when no current is present, is given by [58]:

 is the surface curvature,
is the surface curvature,  is the surface energy, and
is the surface energy, and
 is the elastic strain energy density.
is the elastic strain energy density.

The “double-dot” operator is defined for the tensors  and
and  by
by

and  stands for the components of the
tensor and
stands for the components of the
tensor and  , respectively. The flux parallel to the surface is driven by the chemical
potential of the surface and the EM flux summing up to
, respectively. The flux parallel to the surface is driven by the chemical
potential of the surface and the EM flux summing up to

where the derivatives are taken along the interface and the surface diffuse coefficient  obeys an Arrhenius law
obeys an Arrhenius law

Furthermore, a flux normal to the surface due to a flow of vacancies from the bulk to the surface, reasoned by the difference of the chemical potentials in the bulk and at the surface, is given by

where  controls the rate of vacancy exchange between the bulk and the surface. For
the chemical potential of the vacancies at the surface the negative chemical potential of the surface is taken as given by (3.47). For the bulk the chemical potential is given by:
controls the rate of vacancy exchange between the bulk and the surface. For
the chemical potential of the vacancies at the surface the negative chemical potential of the surface is taken as given by (3.47). For the bulk the chemical potential is given by:

Given these equations and considering the mass conservation the normal velocity of the void surface can be expressed as:

A problem with this model arises from the need to track the interface for every simulation time step, if the FEM is employed as the simulation domains have to be separated in a void region and a metal region and these regions
change in shape. The metal region has to be remeshed for every time step, increasing the simulation complexity. Therefore a model was proposed by Bhate et al. [9] according to Cahn et al. [19], where the boundary between the void
and the metal is not modeled by a sharp interface, but by a thin region called the diffuse interface. To distinguish between the three regions a function called the order parameter  is defined, which has the
property to be 1 in the metal decreasing smoothly in the interface region and being
is defined, which has the
property to be 1 in the metal decreasing smoothly in the interface region and being  in the void region. The free energy functional is
defined by
in the void region. The free energy functional is
defined by

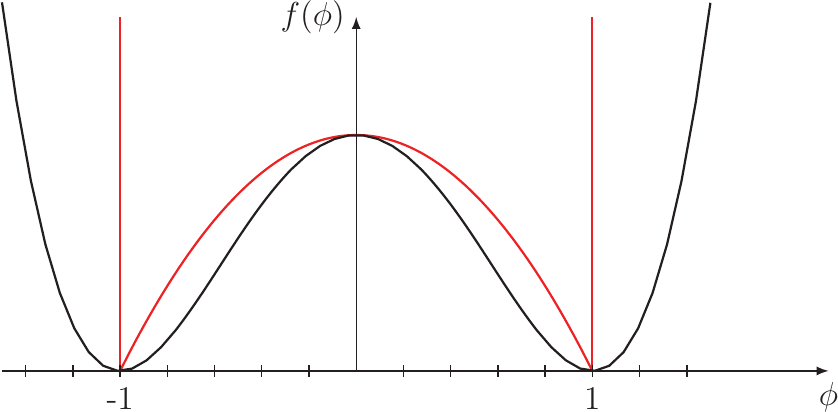
Figure 3.5.: Two shapes proposed for the bulk free energy function  . The quadratic double well potential in black
and the double obstacle potential in red.
. The quadratic double well potential in black
and the double obstacle potential in red.
where  is the elastic strain
energy density given by
is the elastic strain
energy density given by

and is the bulk free energy function. The shape of
this function has to meet the requirements to be positive everywhere and zero for  equaling one or minus one. Two shapes
proposed by different authors are shown in Figure 3.5. The black line is the quadratic double well potential proposed by Mahadevan et al. [99]. The red line is the double obstacle potential proposed by Oono et al. [106]. This
potential was employed by Bhate et al. [9] in a calculation utilizing the finite-difference method, as it reduces the computational effort only to those areas, where the interface is located and the potential has a finite value. The second
term in the energy functional stands for the energy cost due to a gradient of the phase field.
equaling one or minus one. Two shapes
proposed by different authors are shown in Figure 3.5. The black line is the quadratic double well potential proposed by Mahadevan et al. [99]. The red line is the double obstacle potential proposed by Oono et al. [106]. This
potential was employed by Bhate et al. [9] in a calculation utilizing the finite-difference method, as it reduces the computational effort only to those areas, where the interface is located and the potential has a finite value. The second
term in the energy functional stands for the energy cost due to a gradient of the phase field.  is the parameter
which controls the thickness of the interface and has therefore to be chosen carefully, taking into account the meshing diameter and the minimum curvatures of the interface which can occur.
is the parameter
which controls the thickness of the interface and has therefore to be chosen carefully, taking into account the meshing diameter and the minimum curvatures of the interface which can occur.
From this energy functional the chemical potential can be calculated by [36] employing the variational principle [53, 54].

Adopting the flow of the surface (3.50) leads to the phase field flux

and the conservation equation derived from the equation of the normal velocity of the surface

where the flux from the bulk to the interface  as a function of the order
parameter has to vanish outside the interface and is expressed by
as a function of the order
parameter has to vanish outside the interface and is expressed by

As described above the phase field model is an approximation of the sharp interface model for EM. In Section A it is shown, that for the limit, where the interface controlling parameter goes to zero, the
phase field model converges to the sharp interface model [99].
Previous: 3.5 Void Nucleation Top: 3 Models Next: 4 Numerical Implementation