


| | |
Where Section 7.1 described the dynamical system due to Boltzmann’s equation using differential equations, an approach using the integral form of Boltzmann’s equation as introduced in Section 5.6 is provided here. The presented integral equation is approached using the formalism described in Appendix A to approximate solutions. In contrast to the contraction to macroscopic quantities, as described in Section 7.1 this approach makes direct use of the phase space and its geometry by casting trajectories through it. As already outlined in Section 5.4, trajectories alone are no longer sufficient for an adequate description but a mechanism to change between trajectories is also required. The theory outlined in Section 5.6 is first theoretically further refined in order to provide guidelines usable for implementation, reusing components from Section 6.
The continuous model of the evolution of the dynamical system described in Section 5.6, needs only minor adaptations in order to be made accessible to the discrete world of digital computers. The adaptation is briefly outlined to provide a guide of how independent components may be assembled to be applied to a dynamical system.
Since Boltzmann’s equation can be cast in the form of an integral equation, it is possible to construct a resolvent series. Recalling Equation 5.51b:
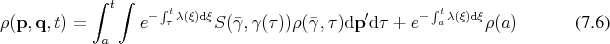
Proceeding as outlined in Appendix A by recursive insertions leads to:
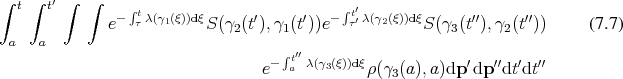
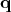 component
using the corresponding parameter, while allowing discontinuous transitions in their
component
using the corresponding parameter, while allowing discontinuous transitions in their 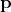 components, as already established in Equation 5.47. Recalling it in adapted form it reads:
components, as already established in Equation 5.47. Recalling it in adapted form it reads:
Another important fact is that the sequence of trajectories here, actually moves back in time from the point of interest to the initial point. Thus the algorithms utilizing this approach directly are aptly called backward algorithms [130].
The trajectories encountered here correspond to the integral curves (Definition 63) of the Hamiltonian vector field (Definition 62). Each trajectory thus is a representative of the Hamiltonian flow (Definition 67), which governs the evolution of the system as a whole. Thus it becomes apparent that the solution is obtained by tracing the trajectories which follow the basic structures which define the geometry of the problem domain.
In the spirit of applying a Monte Carlo approach for the evaluation of the integrals, as described in Section 6.6.2 the factors

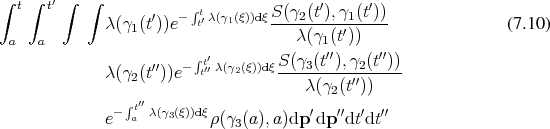
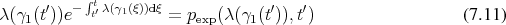

 is not carried
out with the corresponding states. However, such a term can easily by introduced recalling
Equation 5.43
is not carried
out with the corresponding states. However, such a term can easily by introduced recalling
Equation 5.43

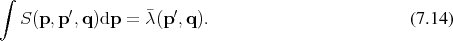
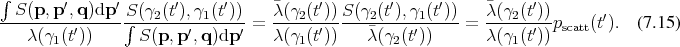

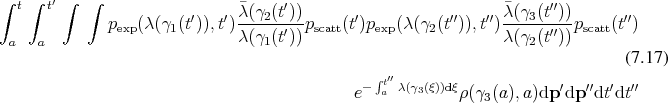
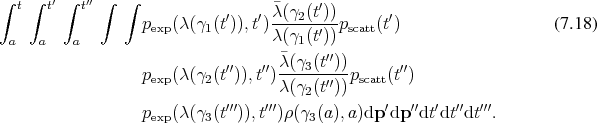
The key components required for the approximation of the described dynamical system can be found from Equation 7.18 to be:
Trajectories have already been introduced in Section 6.5 under the name of curves which shall be specialized for the context of dynamics. While trajectories corresponding to a Hamiltonian flow (Definition 67) fibrate (Definition 39) the whole phase space, only sections are required for the treatment of the dynamical system described by Boltzmann’s equation.
Nevertheless, the sections of the trajectories under investigation are integral curves (Definition 63) of
the Hamiltonian vector field (Definition 62) defined on the phase space. Considering the phase space as
the co-tangent bundle leads to a split of this vector field, into two components which are still tangential
to the curve. However, the two separated components are no longer each a vector field. Where the
component corresponding to 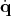 may be immediately identified with a vector field corresponding to the
velocity, the part representing
may be immediately identified with a vector field corresponding to the
velocity, the part representing 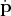 is a tensor field (Definition 65) of type
is a tensor field (Definition 65) of type 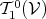 , thus a field of
, thus a field of
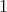 -forms, not a vector field. This field is given by the
-forms, not a vector field. This field is given by the 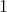 -forms corresponding to the force acting at each
point of the manifold. While the nature of the components seems to have changed when projecting to
the base manifold structures, both fields remain to be an expression of the Hamiltonian which defined
the symplectic geometry.
-forms corresponding to the force acting at each
point of the manifold. While the nature of the components seems to have changed when projecting to
the base manifold structures, both fields remain to be an expression of the Hamiltonian which defined
the symplectic geometry.
The extent of the section of the trajectory traversed corresponds to a random variable using the exponential distribution described in Equation 7.11. A random variable (Definition 98) with the desired properties can be generated from a uniformly distributed random variable by inversion of the distribution function as outlined in Appendix B. Recalling Equation 7.11


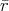 , e.g., from the interval
, e.g., from the interval 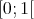 ,
allows to formulate
,
allows to formulate

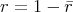 is again a uniformly distributed random variable from the interval
is again a uniformly distributed random variable from the interval 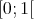 . This
expression can readily be inverted to yield
. This
expression can readily be inverted to yield
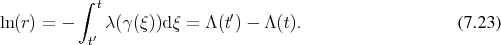
The domain of interest and the quantities associated with it are represented in a discrete manner in the digital computer as described in Section 6.3 and Section 6.4. The discrete nature has repercussions on the validity of the considered trajectory, such that it models only the local flow (Definition 66) instead of the original global flow. The border of validity of any given local trajectory is due to the finite size of the subdivision into cells (Definition 34 and discussed in Section 6.3), which form the base space of the fiber bundle (Definition 40 as implemented in Section 6.4) carrying the quantities which determine the shape of the trajectories.
Since the trajectories are nothing else than curves through phase space, the shape of the the trajectories
is completely defined by the coefficients 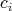 , which were abstractly defined in Section 6.5. Here, they
can be associated to physical quantities.
, which were abstractly defined in Section 6.5. Here, they
can be associated to physical quantities. 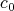 corresponds to the position
corresponds to the position 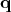 of a particle, while
of a particle, while 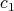 is
linked to
is
linked to 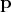 . Finally,
. Finally, 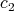 represents
represents 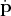 . Thus the geometry of the trajectory is still tightly tied to the
entities of the original symplectic structure.
. Thus the geometry of the trajectory is still tightly tied to the
entities of the original symplectic structure.
The discrete nature makes it necessary to check, if the trajectory leaves the originating cell with any chosen value of the parameter and therefore actually serves as an upper limit for how far the trajectory is traversed, since the trajectory needs to be interrupted in case the parameter carries the new position beyond the initial cell, as already outlined in Section 6.5. Since the trajectories are one-dimensional, it is sufficient to compare the parameters corresponding to the intersection of a trajectory with a current cell’s boundary to the value of the parameter obtained from the random distribution.
Both descriptions of Boltzmann’s equation as provided in Section 4.9 and Section 5.6 acknowledge the defining nature of the scattering term on the right hand side. Thus the scattering term has to be mapped adequately to the realm of digital computing in order to expect the calculated result to be accurate. Furthermore, the scattering term is, together with the geometry defining Hamiltonian, the point of entry for physical modelling into the mathematical structure. Thus, the inclusion of different scattering mechanisms needs not only to be adequate from a mathematical point of view but also must withstand a physicist’s scrutiny.
The scattering kernel 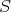 is therefore modelled as being composed from different scattering mechanisms
is therefore modelled as being composed from different scattering mechanisms
 such that
such that

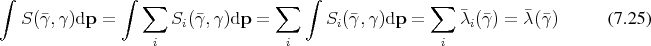
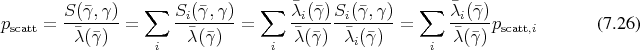
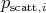 can be related to the total
scattering probability
can be related to the total
scattering probability 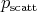 . This relation also allows the selection of a given scattering mechanism by
generating a uniformly distributed random variable
. This relation also allows the selection of a given scattering mechanism by
generating a uniformly distributed random variable 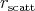 from the interval
from the interval 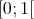 . Since it is possible to
uniquely identify the contribution
. Since it is possible to
uniquely identify the contribution 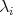 of every scattering mechanism by its index
of every scattering mechanism by its index 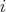 , it is possible to
select a scattering mechanism by choosing an index
, it is possible to
select a scattering mechanism by choosing an index 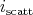 . When choosing this index
. When choosing this index 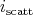 , the
following inequality holds.
, the
following inequality holds.
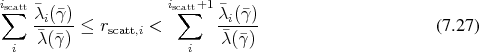
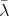 and
expressing it in terms of rates as
and
expressing it in terms of rates as
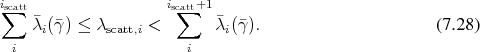
 along with the selection of
the scattering mechanism
along with the selection of
the scattering mechanism 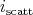 , which determines the beginning of the new trajectory, by means of the
rejection technique as is outlined in Appendix B.
, which determines the beginning of the new trajectory, by means of the
rejection technique as is outlined in Appendix B.
Figure 7.1 provides an illustration of this particular case. At a given point 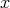 a current rate is chosen
randomly using a distribution from the interval
a current rate is chosen
randomly using a distribution from the interval 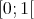 multiplied by an upper bound for all the
scattering. Then the scattering models are evaluated at this point
multiplied by an upper bound for all the
scattering. Then the scattering models are evaluated at this point 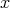 and the individual rates, thusly
calculated are summed in order to determine the interval
and the individual rates, thusly
calculated are summed in order to determine the interval 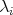 , which contains the current rate. The
scattering mechanism associated with this interval is then chosen as the new state from which the next
trajectory will propagate.
, which contains the current rate. The
scattering mechanism associated with this interval is then chosen as the new state from which the next
trajectory will propagate.
During the simulation the scattering models are evaluated repeatedly in order to choose the next state of the electron under consideration. Therefore high performance is among the requirements placed on the evaluation of the collision model.
Applications implementing scattering mechanisms encountered in the field of solid state electronics, which change the states of electrons have been available quite some time. The foundations for the presented code have been been written in plain ANSI C, where all available scattering mechanisms are implemented as individual functions, which are called subsequently. The scattering models require a variable set of parameters, which leads to non-homogeneous interfaces in the functions representing them. To alleviate this to some extent global variables have been employed completely eliminating any aspirations of data encapsulation and posing a serious problem for attempts for parallelization to take advantage of multi-core CPUs. The code has the very simple and repetitive structure:
Extensions to this code are usually accomplished by copy and paste, which is prone to simple mistakes by oversight, such as failing to change the counter which has to be incremented or calling the incorrect function to update the electron’s state.
Furthermore, at times the need arises to calculate the sum of all the scattering models ( in
Figure 7.1), which is accomplished in a different part of the implementation, thus further opening the
possibility for inconsistencies between the two code paths.
in
Figure 7.1), which is accomplished in a different part of the implementation, thus further opening the
possibility for inconsistencies between the two code paths.
The decision which models to evaluate is performed strictly at run time and it would require significant, if simple, modification of the code to change this at compile time, thus making highly optimized specializations very cumbersome.
The functions calculating the rates and state transitions, however, have been well tested and verified, so that abandoning them would be wasteful.
Object oriented programming directly offers run time polymorphism by means of virtual inheritance. Unfortunately current implementations of inheritance use an intrusive approach for new software components and tightly couple a type and the corresponding operations to the super type. In contrast to object-oriented programming, current applications of generic programming are limited to algorithms using statically and homogeneously typed containers, but offer highly flexible, reusable, and optimizeable software components.
As can be seen, both programming types offer different points of evaluation. Run time polymorphism based on run time concepts [131] tries to combine the virtual inheritance run time modification mechanism with the compile time flexibility and optimization.
Inheritance in the context of run time polymorphism is used to provide an interface template to model the required concept, where the derived class must provide the implementation of the given interface. The following code snippet
therefore introduces a scattering_facade which wraps a scattering_concept part. The virtual inheritance is used to configure the necessary interface parts, in this case rate() and transition(), which have to be implemented by any scattering model. In the given example the state_type is still available for explicit parametrization.
To interface this novel approach a core structure is implemented which wraps the implementations of the scattering models:
By supplying the required parameters at construction time it is possible to homogenize the interface of the operator(). This methodology also allows the continued use of the old data structures in the initial phases of transition, while not being so constrictive as to hamper future developments.
The functions for the state transitions are treated similarly to those for the rate calculation. Both are then fused in a scattering_pack to form the complete scattering model and to ensure consistency of the rate and state transition calculations as can be seen in the following part of code:
The blend of run time and compile time mechanisms allows the storage of all scattering models within a single container, e.g. std::vector, which can be iterated over in order to evaluate them.
For the development of new collision models easy extendability, even without recompilations, is also a highly important issue. This approach allows the addition of scattering models at run time and to expose an interface to an interpreted language such as, e.g., Python [29].
In case a highly optimized version is desired, the run time container (here the std::vector) may be exchanged by a compile time container, which is also readily available from the GSSE and provides the compiler with further opportunities for optimizations at the expense of run time adaptability.
While the described approach initially slightly increases the burden of implementation, due to the fact that wrappers must be provided, it gives a transition path to integrate legacy codes into an up to date frame while at the same time not abandoning the experience associated with it. The invested effort allows to raise the level of abstraction, which in turn allows to increase the benefits obtained from the advances in compiler technologies. This in turn inherently allows an optimization for several platforms without the need for massive human effort, which was needed in previous approaches.
In this particular case, encapsulating the reliance on global variables of the functions implementing the scattering models to the wrapping structures, parallelization efforts are greatly facilitated, which are increasingly important with the continued increase of computing cores per CPU.
Furthermore the results can easily be verified as code parts are gradually moved to newer implementations, the only stringent requirement being link compatibility with C++. This test and verification can be taken a step further in case the original implementation is written in ANSI C, due to the high compatibility of it to C++. It is possible to weave parts of the new implementation into the older code. Providing the opportunity to get a comparison not only of final results, but of all the intermediates as well.
Such swift verification of implementations allows to also speed up the steps necessary to verify calculated results with subsequent or contemporary experiments, which should not be neglected, in order to keep physical models and their numerical representations strongly rooted in reality.
The correct operation of the run time concepts depends on the faultless operation of the compiler being used within the rules set down by the C++ language specification [132]. Under this crucial assumption the correct compilation of the code ensures that the code will behave according to the guarantees introduced by the run time concept. While this provides an indication for correctness it cannot be considered absolute proof, especially since the C++ language and its compilers does not have a special focus on validation as is the case in, e.g., Ada[133][134], which could certainly make a stronger case.
| | |

